Q85.0 Нейрофиброматоз (незлокачественный), МКБ-10
Отредактировано: 11.12.2025
Нейрофиброматоз
Определение
Нейрофиброматозы – группа наследственных заболеваний с аутосомно-доминантным типом наследования, общим клиническим проявлением которых является формирование множественных опухолей в тканях нейроэктодермального происхождения [1].
Классификация
Существует несколько клинически и генетически различных форм нейрофиброматоза. Изначально выделяли:
- нейрофиброматоз 1 типа (НФ1),
- нейрофиброматоз 2 типа (НФ2),
- шванноматозы.
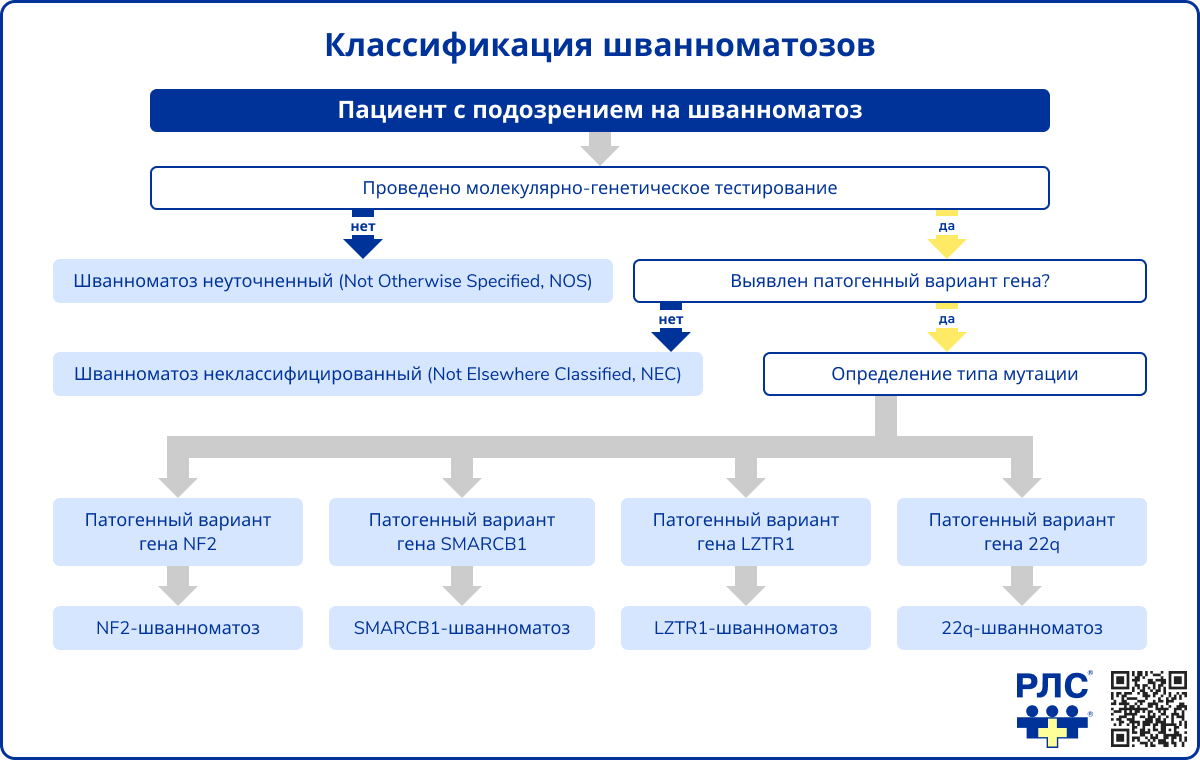
Однако в 2022 году в журнале «Genetics in Medicine» были опубликованы результаты международного консенсуса, по итогам которого НФ2 и шванноматоз были объединены в группу синдромов, характеризующихся повышенным риском развития шванном, и обозначены термином «шванноматозы» [2]. При этом каждый тип шванноматоза классифицируется в соответствии с геном, в котором идентифицируется патогенный вариант. Таким образом, согласно пересмотренной номенклатуре, выделяют:
- Шванноматоз, ассоциированный с патогенным вариантом NF2 (NF2-шванноматоз);
- Шванноматоз, ассоциированный с патогенным вариантом SMARCB1 (SMARCB1-шванноматоз);
- Шванноматоз, ассоциированный с патогенным вариантом LZTR1 (LZTR1-шванноматоз);
- Шванноматоз, ассоциированный с патогенным вариантом в длинном плече 22-й хромосомы (22q-шванноматоз);
- Шванноматоз неуточненный (not otherwise specified, NOS) – к этой группе относятся пациенты с клиническими признаками шванноматоза, не проходившие генетическое тестирование;
- Шванноматоз неклассифицированный (not elsewhere classified, NEC) – к этой группе относятся пациенты с клиническими признаками шванноматоза, у которых не было выявлено патогенного варианта по результатам генетического тестирования.
Нейрофиброматоз 1 типа
Определение
Нейрофиброматоз 1 типа (НФ1), ранее известный как болезнь Реклингхаузена, представляет собой мультисистемное генетическое заболевание и является наиболее распространенной формой нейрофиброматоза [3]. Это заболевание характеризуется аутосомно-доминантным типом наследования и полной пенетрантностью. Полная пенетрантность означает, что если у человека есть патогенная мутация в гене NF1, то клинические признаки болезни проявятся у 100% носителей в течение жизни, однако вариабельная экспрессивность обусловливает различную тяжесть симптомов – от легких форм до тяжелых проявлений заболевания – даже у лиц из одной семьи. Около половины случаев НФ1 вызваны de novo мутациями в гене NF1, преимущественно в хромосомах отцовского происхождения [4].
Характерными признаками НФ1 служат множественные пятна цвета «кофе с молоком» (café-au-lait) и нейрофибромы. Если клинические проявления ограничены одним участком тела из-за соматического мозаицизма патогенного варианта в гене нейрофибромина 1, состояние обозначается как сегментарный НФ1 [5].
Частота встречаемости НФ1 составляет приблизительно 1:3000 новорожденных и не зависит от этнической и расовой принадлежности [6]. Частота встречаемости сегментарного НФ1 точно неизвестна, оценивается как 1:36 000-1:40 000 человек [7].
Ожидаемая продолжительность жизни пациентов с НФ1 снижается в среднем на 8-15 лет [8]. Исследование в США показало, что средний возраст смерти пациентов с НФ1 составляет 54,4 года, что значительно ниже средних популяционных показателей (70,1 лет) [9]. Главной причиной смерти пациентов молодого возраста (до 30 лет) являются злокачественные опухоли, в частности:
- злокачественные опухоли оболочек периферических нервов [10],
- глиомы зрительного пути [11],
- рабдомиосаркомы [12, 13],
- феохромоцитомы [8, 14].
Этиология
НФ1 развивается из-за патогенных аберраций в гене NF1, расположенном в хромосомной области 17q11.2 [15, 16]. Описано более 3000 патогенных вариантов NF1, включая нонсенс- и миссенс-мутации, делеции, инсерции и интронные варианты, нарушающие сплайсинг. Широкий спектр мутаций обусловлен в том числе большим размером гена NF1, охватывающего ≈350 000 пар нуклеотидов, при этом:
- 85-90% приходится на точечные мутации,
- 5-10% – на микроделеции,
- 2% – на делеции или дупликации отдельных экзонов.
Появление преждевременных стоп-кодонов, являющееся результатом примерно 80% мутаций, влечет за собой синтез укороченных нефункциональных белков либо отсутствие полноценного транскрипта. Характерна высокая частота de novo мутаций (≈50% случаев), что связывают с наличием Alu-повторов, способствующих неравному рекомбинационному кроссинговеру [17].
Исследования выявили «генотип-фенотипические корреляции» для патогенных вариантов NF1 и ассоциированных клинических проявлений [18, 19]. Например, кластеризация мутаций в 5'-концевой области гена NF1 ассоциируется с повышенным риском развития глиом зрительного нерва [20]. Таким образом, детальный молекулярно-генетический анализ позволяет выделить пациентов, требующих более частого и целенаправленного офтальмологического наблюдения из-за выявленных рисков.
Ген NF1 кодирует нейрофибромин – онкосупрессорный белок, регулирующий клеточную пролиферацию. Нейрофибромин относится к семейству GAP-белков (GTPase-activating proteins) и ускоряет гидролиз активной формы белка Ras-ГТФ до неактивной Ras-ГДФ, тем самым регулируя сигнальные каскады, опосредуемые белками семейства Ras1 [21, 22]. Белки Ras связаны с рядом сигнальных путей, способствующих активации пролиферативной активности клеток, включая:
- сигнальный путь SCF2 /c-kit3 ,
- сигнальный путь mTOR4 ,
- сигнальный путь MAPK5 .
Таким образом, потеря или снижение функции нейрофибромина приводит к гиперактивации Ras-зависимых путей и, как следствие, к неконтролируемой пролиферации клеток, что лежит в основе повышенного риска развития опухолей при НФ1, включая злокачественные новообразования.
Для НФ1 характерна практически полная пенетрантность к взрослому возрасту, однако отмечается выраженная внутрисемейная вариабельность фенотипа. Это, вероятно, обусловлено следующими факторами:
- широким спектром возможных мутаций в гене NF1,
- аллельной гетерогенностью,
- эпистазом,
- эпигенетическими факторами.
Возможен соматический мозаицизм из-за постзиготической мутации в гене NF1 [7]. Это приводит к тому, что одни клетки имеют два полностью функциональных гена NF1, а другие содержат патогенный вариант одной копии гена NF1. Если мозаицизм затрагивает и соматические, и половые (гонадные) клетки, при передаче патогенного варианта потомку последний унаследует вариант в геномном (нераспределенном) состоянии, и клинические проявления будут генерализованными.
Концепция мозаицизма объясняет разнообразие клинических проявлений мозаичных форм НФ1, которое может определяться временем возникновения мутации NF1, типом пораженной клеточной линии и жизнеспособностью мутировавших клеток [23, 24]. Формы НФ1, ассоциированные с мозаицизмом, включают:
- Сегментарный (локализованный) НФ1 – клинические проявления ограничены одним или несколькими сегментами тела, обычно на одной стороне.
- Генерализованный НФ1 – фенотипически схож с классическим НФ1, но при анализе крови мутация может не обнаруживаться; как правило, течение более легкое.
- Гонадный мозаицизм – патогенный вариант присутствует только в половых клетках.
Наиболее распространенным среди перечисленных вариантов является сегментарный фенотип НФ1. При сегментарном НФ1 мутация возникает на поздних стадиях эмбриогенеза, что обусловливает локализованный характер заболевания.
Патогенез
Патогномоничным проявлением НФ1 является нейрофиброма – опухоль периферического нерва смешанного состава (шванновские клетки, фибробласты, эндотелиальные клетки, тучные клетки, макрофаги, нейроны и внеклеточный матрикс) [1]. Ключевую роль в развитии нейрофибром играют шванновские клетки, которые считаются инициирующим звеном в развитии опухоли. Это подтверждается тем, что именно в них происходит утрата второй функциональной копии гена NF1 (так называемый «второй удар» согласно модели Кнудсона). Системный характер поражений при НФ1 объясняется тем, что мутация в гене NF1 нарушает развитие не только шванновских клеток, но и многих других производных нейрального гребня. Это приводит к широкому спектру проявлений, затрагивающих кожу, кости, периферические нервы и другие ткани.
Клиническая картина
Кожные проявления НФ1, как правило, манифестируют в раннем детском возрасте и характеризуются тенденцией к сохранению и прогрессированию в течение жизни.
- Пятна цвета «кофе с молоком» (café-au-lait macules) встречаются в 95% случаев. Они представляют собой плоские, однородные по структуре пятна от светло-бежевого до темно-коричневого цвета, округлой или овальной формы, с ровным контуром и четкими границами. Появляются обычно на первом году жизни, характерно увеличение размеров пятен пропорционально росту ребенка. При морфологическом исследовании выявляется диффузное скопление меланоцитов в сосочковом слое дермы, содержащих гранулы меланина в цитоплазме.
- Веснушки (лентиго) в подмышечных или паховой областях представляют собой мелкие (до 2 мм) пигментированные макулы, расположенные в кожных складках. Появляются позднее пятен цвета «кофе с молоком» – обычно в 3-5 лет.
Офтальмологические симптомы при НФ1 включают как состояния, непосредственно связанные с поражением глазного яблока и зрительного пути, так и вторичные изменения. Характерные находки включают:
- Узелки Лиша – пигментированные гамартомы радужной оболочки, выявляемые при осмотре на щелевой лампе как мелкие желто-коричневые куполообразные папулы. Они специфичны для НФ1, чаще развиваются после 6 лет;
- Аномалии сосудистой оболочки глаза.
Опухолевые проявления НФ1 многообразны – от доброкачественных образований кожи до злокачественных поражений нервной системы и внутренних органов.
- Кожные и подкожные нейрофибромы присутствуют почти у всех пациентов, происходят из терминалей кожных нервов. Они обычно появляются в пубертатном периоде, их количество увеличивается с возрастом. Эти образования являются доброкачественными, характеризуются крайне низким риском злокачественной трансформации, однако часто представляют для пациента косметическую проблему.
- Плексиформные нейрофибромы возникают примерно у 30% пациентов, развиваются из нервных сплетений. Термин «плексиформная» происходит от лат. «plexus» – сплетение и «formis» – подобный. Плексиформные нейрофибромы характеризуются развитой васкуляризацией и инфильтративным ростом вдоль нервных волокон с вовлечением множества нервных пучков. Они формируются чаще у детей младше 5 лет, отличаются быстрым ростом, приводящим к компрессии и деформации окружающих структур. Эти образования могут сдавливать дыхательные пути или спинной мозг, а также трансформироваться в злокачественные шванномы периферических нервов. Наиболее частым признаком злокачественной трансформации плексиформной нейрофибромы является появление болезненности и стремительное увеличение образования в размерах.
- Атипичные нейрофибромы или атипичные нейрофиброматозные новообразования с неопределенным биологическим потенциалом представляют собой образования, имеющие признаки атипии при гистологическом исследовании, включая повышенную митотическую активность, утрату архитектоники ткани и высокую клеточность [26, 27]. Они могут накапливать 18-фтордезоксиглюкозу при ПЭТ-исследовании. Атипичные нейрофибромы характеризуются гомозиготной делецией гена-ингибитора циклин-зависимой киназы 2A/B (CDKN2A/B) в дополнение к потере обоих аллелей NF1, и рассматриваются как предраковые образования, поэтому требуют повышенного внимания, тщательного клинического мониторинга и регулярных МРТ-исследований для динамической оценки размеров и морфологических характеристик образований [28, 29].
- Глиома зрительного пути представляет собой пилоцитарную астроцитому – доброкачественную, медленно растущую опухоль, которая поражает зрительный нерв, хиазму или зрительные тракты. Она диагностируется у 15-20% пациентов с НФ1, обычно манифестирует к 6 годам жизни. Клиническое течение может быть бессимптомным либо сопровождаться следующими нарушениями: снижение остроты зрения, потеря цветовосприятия, экзофтальм, нарушение полей зрения, преждевременное половое созревание (если опухоль затрагивает гипоталамус). Наиболее значимыми факторами риска прогрессирующей потери зрения при глиоме зрительного пути являются:
- возраст младше 2 лет,
- женский пол,
- поражение захиазмальных отделов зрительного пути.
- Злокачественная опухоль из оболочек периферических нервов (ЗООПН) является высокоагрессивным вариантом саркомы мягких тканей. Пожизненный риск развития ЗООПН у пациентов с НФ1 составляет от 8 до 13%, риск возрастает с увеличением возраста пациента. В большинстве случаев ЗООПН развиваются из уже существующих плексиформных или атипичных нейрофибром. К клиническим признакам, указывающим на злокачественную трансформацию нейрофибромы, относят:
- появление сильной и постоянной боли в области нейрофибромы,
- изменение консистенции опухоли с мягкой на плотную,
- быстрый рост узла в пределах существующей плексиформной нейрофибромы.
- Кроме перечисленных выше, у пациентов с НФ1 встречаются и другие опухоли, среди которых:
- глиомы ствола головного мозга,
- саркомы мягких тканей,
- гломусные опухоли,
- лимфопролиферативные заболевания,
- рак молочной железы,
- феохромоцитома,
- гастроинтестинальные стромальные опухоли.
Для НФ1 характерен специфический спектр ортопедических нарушений, обусловленных патологией опорно-двигательного аппарата:
- дисплазия длинных трубчатых костей,
- псевдоартроз,
- низкорослость,
- сколиотическая деформация позвоночника,
- дисплазия крыла клиновидной кости,
- остеопороз.
Неврологические и когнитивные нарушения при НФ1 включают как структурно обусловленные, так и функциональные расстройства.
- Когнитивные нарушения выявляются примерно у 50% пациентов с НФ1. Они могут быть связаны с осложнениями НФ1 (например, эпилептической энцефалопатией) или быть результатом ассоциированных заболеваний (например, расстройств аутистического спектра). Синдром дефицита внимания и гиперактивности (СДВГ) диагностируется у 30-40% детей с НФ1. Часто встречаются проблемы с артикуляцией, также могут наблюдаться нарушения слуховой обработки информации и снижение речевой дискриминации (трудности с различением звуков речи).
- Макроцефалия может быть как относительной (непропорционально большая голова по отношению к росту), так и абсолютной (окружность головы превышает 98-ю перцентиль). Основная причина – увеличенный объем мозга (мегалэнцефалия). В редких случаях макроцефалия может являться следствием гидроцефалии, вызванной стенозом водопровода мозга.
- Эпилепсия часто является проявлением НФ1. Частота встречаемости судорог у пациентов с НФ1 примерно в два раза выше, чем в общей популяции, и составляет около 4-6%. Приступы могут быть любого типа (фокальные, генерализованные) и манифестировать в любом возрасте. В большинстве случаев судороги не связаны с объемным образованием в головном мозге, однако возникновение фокальных приступов может быть обусловлено внутричерепной опухолью. В 30% случаев эпилепсия не поддается лечению противосудорожными препаратами, что может быть ассоциировано с выраженными когнитивными нарушениями.
- У пациентов с НФ1 нередко развивается периферическая нейропатия. Причина заключается в компрессии нервов (до 4% случаев) или компрессии спинномозговых корешков (до 3% случаев). Периферическая нейропатия может возникать и без видимой компрессии нерва, в том числе протекать в форме поражения мелких нервных волокон (small fiber neuropathy), что проявляется болью, жжением и нарушениями чувствительности.
К другим проявлениям НФ1 относятся врожденные пороки сердца. Наиболее частыми являются:
- стеноз клапана легочной артерии,
- дефекты межпредсердной и межжелудочковой перегородок,
- коарктация аорты,
- тетрада Фалло,
- пролапс митрального клапана,
- гипертрофическая кардиомиопатия.
Артериальная гипертензия является частым клиническим проявлением НФ1 у взрослых, реже может дебютировать в детском возрасте. В большинстве случаев гипертензия считается первичной (эссенциальной), однако сосудистые поражения, ведущие к реноваскулярной гипертензии, у пациентов с НФ1 встречаются значительно чаще. Гораздо менее распространенной, но крайне важной причиной гипертензии при НФ1 является феохромоцитома.
Согласно данным исследований, при НФ1 повышается риск развития заболеваний желудочно-кишечного тракта:
В клинической картине НФ1 могут встречаться легочные поражения, включая:
- легочную гипертензию,
- кисты легких,
- фиброз легочной ткани,
- интерстициальные поражения легких,
- эмфизематозные изменения.
Клиническая картина НФ1 отличается выраженной вариабельностью, при этом прослеживается корреляция между возрастом пациента и манифестацией наиболее характерных симптомов.

Источник: Bruce R Korf, Mina Lobbous, Laura K Metrock. Neurofibromatosis type 1 (NF1): Pathogenesis, clinical features, and diagnosis - UpToDate [Электронный ресурс]. URL: https://www.uptodate.com/contents/neurofibromatosis-type-1-nf1-pathogenesis-clinical-features-and-diagnosis (дата обращения: 28.07.2025).
Методы диагностики
При обследовании ребенка с подозрением на НФ1 целесообразно выполнить следующие мероприятия:
- собрать подробный семейный анамнез с упором на признаки НФ1 у родных первой линии,
- при возможности провести клинический осмотр биологических родителей и сиблингов для выявления характерной фенотипической симптоматики НФ1.
Цель физикального обследования – выявление системных и локальных проявлений НФ1 и оценка сопутствующей патологии. При обследовании рекомендуется провести:
- общий осмотр кожных покровов,
- измерение окружности головы, измерение роста и веса с оценкой скорости роста,
- общий ортопедический осмотр (осмотр позвоночника, грудной клетки, конечностей для исключения деформации),
- общую оценку неврологического, эндокринологического, офтальмологического статусов, когнитивных способностей,
- пальпацию мягких тканей, осмотр и перкуссию зон с предполагаемыми новообразованиями,
- измерение артериального давления на периферических артериях.
Ускоренный линейный рост может выступать ранним маркером преждевременного полового созревания, поэтому рекомендуется регулярный контроль роста с использованием возрастно-половых перцентильных таблиц и документированием скорости роста при динамическом наблюдении детей с НФ1.
Рекомендуемые лабораторные исследования направлены на оценку общего состояния пациента и включают:
- биохимический анализ крови,
- общий анализ крови,
- общий анализ мочи.
Молекулярно-генетическое тестирование показано для подтверждения диагноза в сомнительных клинических ситуациях и для целевого скрининга родственников. Исследование показано при пренатальной или преимплантационной диагностике. Выявление мутации в гене NF1 при молекулярно-генетическом тестировании не позволяет предсказать тяжесть течения заболевания или риск осложнений, за исключением некоторых генотипов, ассоциированных с известными фенотипическими особенностями.
Инструментальная диагностика при подозрении на НФ1 включает следующие методы:
- магнитно-резонансная томография (по показаниям),
- электрокардиография (ЭКГ),
- офтальмологический осмотр с использованием щелевой лампы.
Показания к биопсии опухолевых и опухолеподобных образований у пациентов с НФ1 включают:
- подозрение на злокачественную трансформацию,
- отсутствие четкой клинической картины,
- настораживающие изменения на МРТ, ПЭТ/КТ,
- стремительное увеличение размеров стабильной плексиформной нейрофибромы или появление стойкого болевого синдрома.
В 1988 году Национальный институт здоровья США (National Institutes of Health, NIH) разработал критерии для постановки диагноза НФ1 [30]. Согласно актуальной редакции от 2021 года, диагноз НФ1 может быть выставлен при соблюдении одного из следующих условий [23]:
- Наличие двух и более диагностических критериев у пациента, чьи родители не страдают НФ1;
- Наличие одного и более диагностических критериев у пациента, у которого хотя бы один из родителей болен НФ1.

Источник: Legius E. и др. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: an international consensus recommendation // Genetics in Medicine. Elsevier B.V., 2021. Vol. 23, № 8. P. 1506–1513.
Чувствительность и специфичность диагностических критериев у пациентов раннего детского возраста ниже, поскольку многие фенотипические признаки НФ1 формируются позднее. Поэтому пациенты раннего детского возраста с единственным выявленным диагностическим критерием и отсутствием семейного анамнеза НФ1 подлежат динамическому наблюдению для своевременного выявления других возможных проявлений и коррекции терапии при необходимости [31]. Как правило, окончательный клинический диагноз НФ1 устанавливают к 3-4 годам жизни.
Для мозаичных форм НФ1 разработаны отдельные диагностические критерии. Диагноз считается подтвержденным, если у пациента присутствует хотя бы один из нижеперечисленных критериев.

Источник: Legius E. и др. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: an international consensus recommendation // Genetics in Medicine. Elsevier B.V., 2021. Vol. 23, № 8. P. 1506–1513.
Таким образом, оптимизация диагностики и ведения пациентов с подозрением на НФ1 требует комплексного междисциплинарного подхода. Обследование целесообразно проводить с привлечением команды специалистов, включающей педиатра, невролога, генетика, офтальмолога, дерматолога и ортопеда-травматолога.
Задачами мультидисциплинарной команды врачей являются:
- точная верификация диагноза на основании международных диагностических критериев,
- раннее выявление поддающихся лечению осложнений,
- разработка индивидуального плана наблюдения и профилактики осложнений,
- своевременное направление к необходимым узким специалистам.
Дифференциальная диагностика
Ряд наследственных заболеваний характеризуется клинической картиной, сходной с проявлениями НФ1, однако требует иных подходов к ведению пациентов. Поэтому проведение расширенной дифференциальной диагностики является необходимым условием, поскольку установление точного нозологического диагноза позволяет не только адекватно оценить риски специфических осложнений, но и разработать персонализированный план наблюдения. Ключевую роль в различении этих патологий играет молекулярно-генетическое тестирование. Основными заболеваниями для дифференциальной диагностики при подозрении на НФ1 являются [25, 32-35]:
- синдром Легиуса,
- синдром дефицита репарации ошибочно спаренных нуклеотидов6 ,
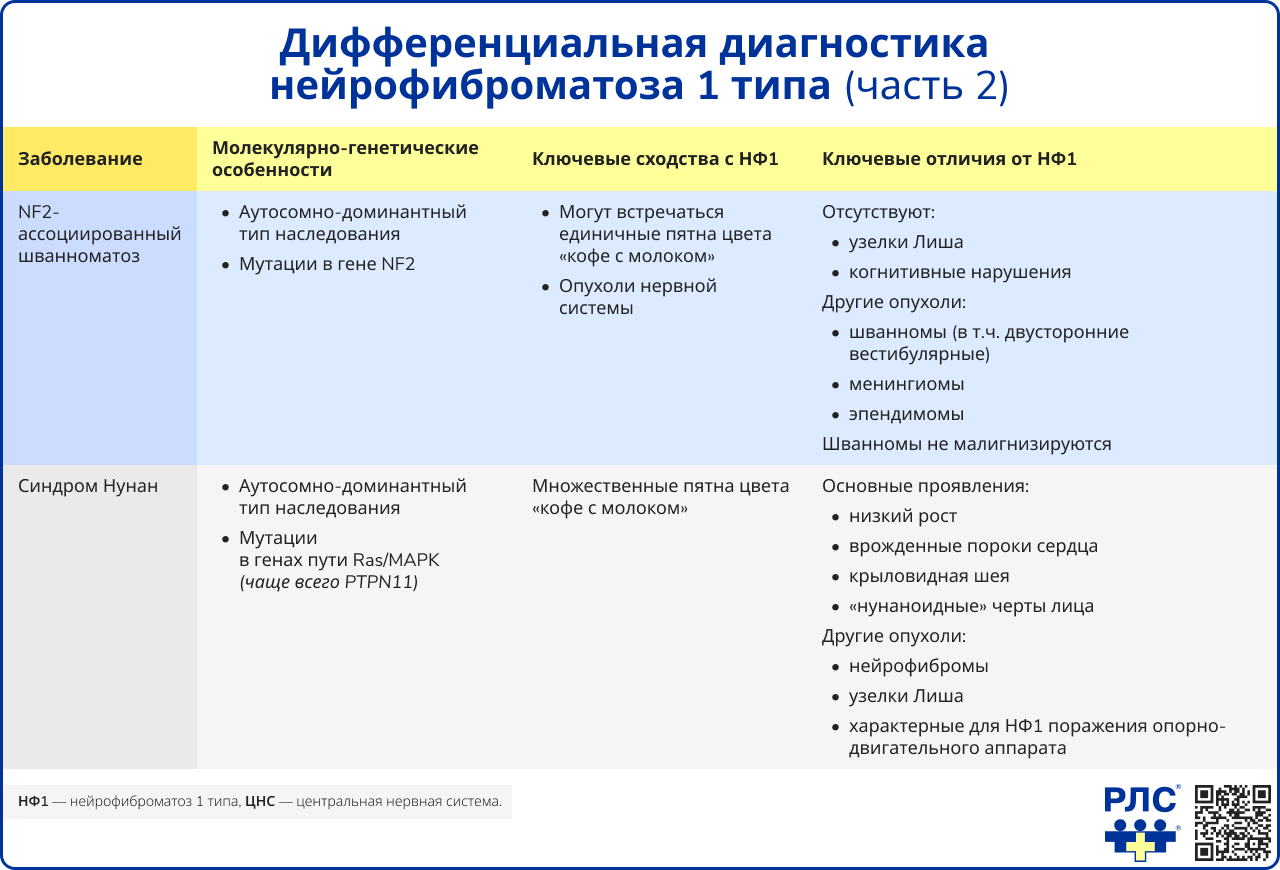
- NF2-ассоциированный шванноматоз (NF2-шванноматоз),
- синдром Нунан.

Лечение
Поскольку НФ1 представляет собой мультисистемное заболевание с высокой вариабельностью клинических проявлений, пациентам требуется пожизненное наблюдение у медицинских специалистов различного профиля. Гетерогенность клинической картины НФ1 исключает единый терапевтический подход и обусловливает необходимость составления индивидуального плана ведения для каждого пациента. Основу лечения составляет проактивный мониторинг, направленный на раннее выявление и лечение специфичных для возраста осложнений по мере их возникновения. Такой подход позволяет улучшить качество жизни пациентов и прогноз заболевания.
Удаление кожных и подкожных нейрофибром проводится только при наличии конкретных показаний, таких как болевой синдром, кровотечение, нарушение функции органа или беспокоящий пациента косметический дефект [31]. Для удаления доступны различные методы, включая хирургическое иссечение, лазерную деструкцию и электродесикацию.
При зуде у пациента рекомендуется назначение лекарственных средств, применяемых для лечения нейропатической боли, например, габапентина. Терапия зуда антигистаминными препаратами в большинстве случаев неэффективна.
Плексиформные нейрофибромы характеризуются такими особенностями, как инфильтративный рост, частое вовлечение нервных стволов и интенсивная васкуляризация. Эти свойства обусловливают высокий риск развития множественных осложнений, включая хронический болевой синдром, моторную дисфункцию (парезы, параличи) и при определенной локализации – потерю зрения или слуха. Наиболее грозным осложнением является малигнизация плексиформных нейрофибром с трансформацией в злокачественную опухоль оболочки периферических нервов, что значительно ухудшает прогноз.
Терапия плексиформных нейрофибром традиционно сопряжена с трудностями [36, 37]. Хирургическое вмешательство часто носит паллиативный характер и ограничивается:
- Циторедукцией (частичным удалением для уменьшения опухолевой массы) объемных образований для устранения жизнеугрожающих состояний, таких как компрессия спинного мозга или дыхательных путей;
- Удалением крупного мягкотканного компонента для уменьшения функциональных нарушений или достижения приемлемого косметического эффекта.
Радикальная резекция часто невозможна из-за инфильтративного роста опухоли в окружающие ткани и высокой васкуляризации, что сопряжено с риском интраоперационных кровотечений.
Достижением в лечении неоперабельных плексиформных нейрофибром стало одобрение в апреле 2020 года Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США7 селуметиниба – селективного ингибитора MEK1/2 [38]. Показанием к приему этого препарата является лечение педиатрических пациентов в возрасте от трех лет и старше с симптоматическими или прогрессирующими неоперабельными плексиформными нейрофибромами, ассоциированными с НФ1. В настоящее время продолжаются клинические исследования других таргетных препаратов, что открывает новые перспективы для терапии НФ1 [39-41].
Тактика лечения различных опухолей, ассоциированных с НФ1, зависит от их типа, влияния на прилегающие ткани и сопутствующих осложнений. При планировании лечения пациентов с НФ1 и опухолями ЦНС одной из основных задач является ограничение применения лучевой терапии из-за риска развития вторичных злокачественных новообразований ЦНС и сосудистых осложнений. В настоящее время отсутствуют данные о том, что конкретные мутации в гене NF1 ассоциированы с различной чувствительностью к радио- или химиотерапии или позволяют прогнозировать ответ на лечение.


Подходы к лечению неврологических нарушений при НФ1 включают комплекс мероприятий, направленных на коррекцию конкретных проявлений.
- При наличии когнитивного дефицита и трудностей обучения рекомендуется предоставление академической поддержки и организация коррекционных занятий [31, 42]. При наличии нарушений речи и моторики, таких как проблемы с равновесием и походкой, показаны занятия с логопедом, эрготерапия и лечебная физкультура.
- Тактика лечения судорожных приступов у пациентов с НФ1 не отличается от таковой при эпилепсии иной этиологии и подбирается индивидуально неврологом.
- Лечение периферической нейропатии включает, по возможности, устранение причины (например, декомпрессию нерва при его сдавлении опухолью), а также симптоматическую терапию, направленную на облегчение боли и парестезий.
Коррекция нарушений со стороны опорно-двигательной системы требует специализированного ортопедического подхода.
- Дисплазия большеберцовой и других длинных костей, а также сколиоз часто требуют ортопедического вмешательства, включая ношение корсета или хирургическую коррекцию.
- Остеопороз при НФ1 может демонстрировать резистентность к стандартной терапии. Пациентам с дефицитом витамина D показано обязательное восполнение дефицита, в настоящее время изучается целесообразность раннего назначения бисфосфонатов.
- В ряде случаев при псевдоартрозе большеберцовой кости для улучшения мобильности рассматривается ампутация голени с последующим протезированием [43]. Ведение таких сложных случаев целесообразно осуществлять силами мультидисциплинарной ортопедической команды, обладающей опытом работы с НФ1.
Ведение других проявлений НФ1 также требует внимания.
- Многие пациенты, особенно подростки, сталкиваются с психосоциальными проблемами и нарушениями самооценки, обусловленными видимыми проявлениями заболевания, что следует рассматривать как показание для консультации психолога или психотерапевта.
- Тактика ведения артериальной гипертензии зависит от этиологии: эссенциальная гипертензия лечится по стандартным протоколам, в то время как вазоренальная гипертензия или гипертензия, ассоциированная с феохромоцитомой, требуют специфического вмешательства, направленного на устранение причины (ангиопластика, удаление опухоли).
- Редкие, но потенциально опасные легочные осложнения, такие как нейрофибромы средостения, кисты легких, эмфизема и фиброз, лечатся по общепринятым клиническим протоколам.
Шванноматоз
Определение
Шванноматозы – группа наследственных заболеваний, обусловленных патогенными вариантами генов-онкосупрессоров на хромосоме 22 и характеризующихся развитием множественных доброкачественных новообразований центральной и периферической нервной системы [44].
Механизм наследования шванноматозов – аутосомно-доминантный, пенетрантность различается для разных генетических подтипов [45]:
- При NF2-ассоциированном шванноматозе пенетрантность составляет 100%;
- При шванноматозах, обусловленных патогенными вариантами генов LZTR1 и SMARCB1, пенетрантность неполная. Семейный анамнез обнаруживается менее чем у 20% пациентов с верифицированным диагнозом.
NF2-шванноматоз – наиболее частый подтип; ориентировочная распространенность оценивается в пределах порядка 1:50 000 новорожденных [6]. LZTR1- и SMARCB1-шванноматозы встречаются реже; распространенность составляет приблизительно 1:126 000 [46]. Существующие оценки, вероятно, занижены из-за трудностей клинической и молекулярно-генетической верификации диагноза.
Этиология и патогенез
Молекулярные механизмы шванноматозов многоступенчаты и преимущественно связаны с инактивацией генов-онкосупрессоров на хромосоме 22.
NF2-шванноматоз возникает из-за инактивирующих мутаций в гене NF2, локализованном на хромосоме 22q12.2 [47, 48]. Этот ген кодирует белок мерлин (также известный как шванномин), который выполняет роль супрессора опухолевого роста.
Формирование опухолей при NF2-шванноматозе соответствует классической двухударной (two-hit) модели Кнудсона:
- Первый «удар» – герминальная инактивация одной аллели гена NF2;
- Второй «удар» – соматическая инактивация оставшейся аллели на гомологичной хромосоме.
Примерно 25-30% случаев NF2 соответствуют мозаичным формам, возникающим из-за постзиготических мутаций. Эти случаи характеризуются более легким течением и ограниченным распространением опухолей.
LZTR1-шванноматоз обусловлен мутациями в гене LZTR18 , расположенном в области 22q11.21 [49]. LZTR1 является компонентом CUL3-убиквитинлигазного комплекса9 и участвует в:
- регуляции пролиферации клеток через контроль деградации ключевых сигнальных молекул,
- модуляции активности Ras-ГТФазы,
- контроле клеточной дифференцировки [50].
SMARCB1-шванноматоз вызван мутациями в гене SMARCB110 , расположенном на хромосоме 22q11.23 [51]. Белок, который кодируется геном SMARCB1, является ключевым компонентом сложного белкового комплекса, который участвует в:
- регуляции транскрипции через изменение структуры хроматина,
- контроле клеточного цикла,
- поддержании целостности генома.
Клиническая картина
Клиническая картина у пациентов с различными типами шванноматозов отличается выраженной клинической вариабельностью [52]. К наиболее частым клиническим проявлениям NF2-шванноматоза относятся следующие:
- Поражения нервной системы: характеризуются развитием специфических опухолевых образований.
- Двусторонние вестибулярные шванномы, обычно манифестирующие к 30 годам, встречаются у 90-95% пациентов.
- Шванномы других черепных нервов выявляются у 24-51% больных, внутричерепные менингиомы – у 45-77%.
- Опухоли спинного мозга (как интрамедуллярные, так и экстрамедуллярные) диагностируются у 63-90% пациентов, а периферическая нейропатия – до 66% случаев.
- Зрительные нарушения: наиболее распространенным проявлением является катаракта (60-81%), за которой следуют эпиретинальная мембрана (12-40%) и гамартомы сетчатки (6-22%)
- Кожные поражения: включают опухоли кожи (59-68%), кожные бляшки (41-48%) и опухоли подкожной жировой клетчатки (43-48%).
Для LZTR1- и SMARCB1-шванноматозов характерно развитие множественных шванном (за исключением внутрикожных) [46, 53]. Шванномы обычно поражают периферические и спинномозговые нервы. Симптомы чаще всего манифестируют в 20-40 лет. Наиболее частые первичные проявления – это локализованная или диффузная боль либо бессимптомное объемное образование. Менингиомы чаще ассоциированы со SMARCB1-шванноматозом, при LZTR1-шванноматозе менингиомы регистрируются значительно реже [54]. Существует риск злокачественного перерождения образований, особенно у лиц с SMARCB1-шванноматозом.
Диагностика
На первом этапе проводится детальный сбор жалоб, клинического и семейного анамнеза.
Физикальное обследование предусматривает:
- общий осмотр кожных покровов,
- общую оценку неврологического,
- эндокринологического и офтальмологического статусов,
- пальпацию и перкуссию возможных образований.
Лабораторная диагностика включает:
- выполнение биохимического и общего анализа крови, общего анализа мочи,
- проведение молекулярно-генетического тестирования.
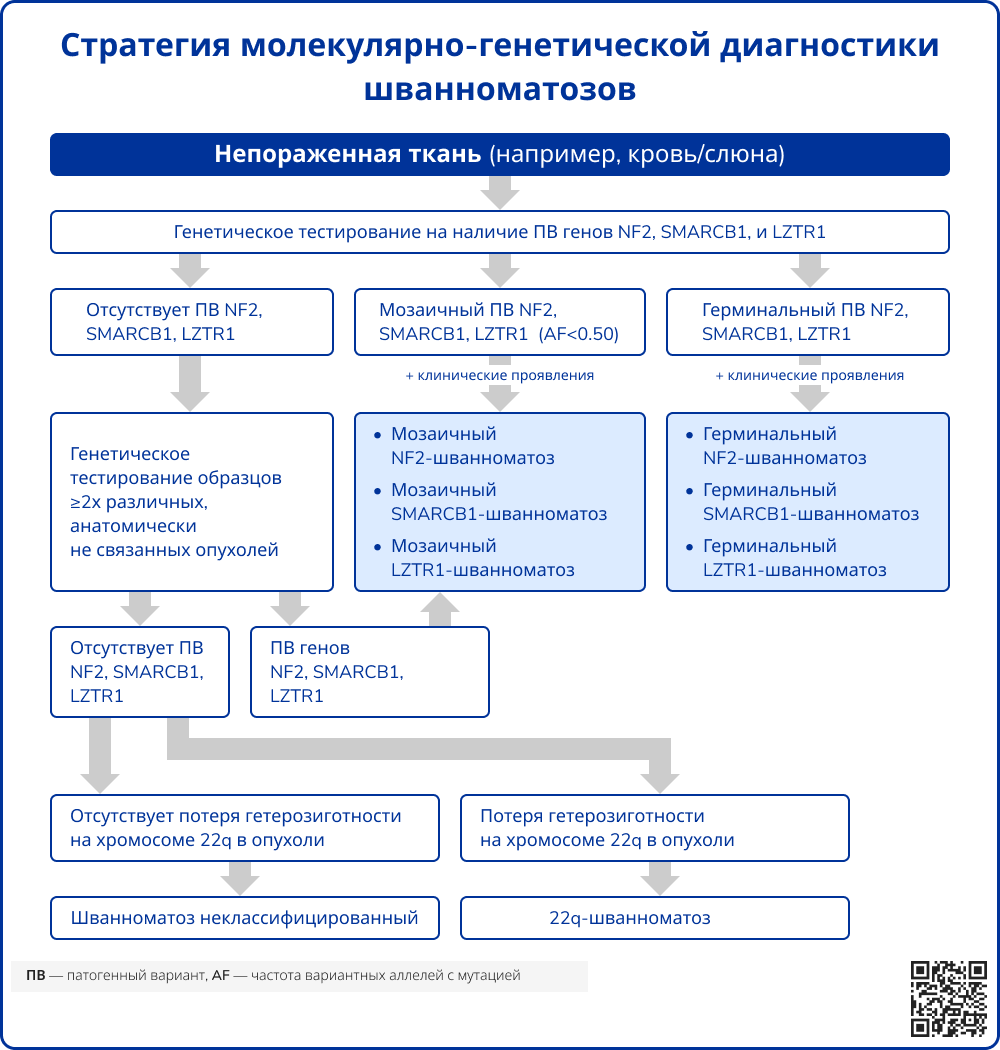
Генетическое тестирование рекомендуется всем пациентам с подозрением на шванноматоз, однако для верификации диагноза NF2-шванноматоза у пациентов, соответствующих установленным клиническим критериям, оно не является обязательным.
Инструментальные методы исследования включают:
- магнитно-резонансную томографию головного мозга и позвоночного столба с контрастным усилением, которая показана всем пациентам с подозрением на шванноматоз,
- аудиометрию,
- офтальмологический осмотр с использованием щелевой лампы.
Постановка диагноза различных видов шванноматозов осуществляется на основании диагностических критериев [2].


Источник: Plotkin S.R. и др. Updated diagnostic criteria and nomenclature for neurofibromatosis type 2 and schwannomatosis: An international consensus recommendation // Genetics in Medicine. Elsevier B.V., 2022. Т. 24, № 9. С. 1967–1977.
Источник: Plotkin S.R. и др. Updated diagnostic criteria and nomenclature for neurofibromatosis type 2 and schwannomatosis: An international consensus recommendation // Genetics in Medicine. Elsevier B.V., 2022. Т. 24, № 9. С. 1967–1977.
Лечение
Терапия шванноматозов требует мультидисциплинарного подхода из-за полиморфизма клинических проявлений. Лечение является преимущественно симптоматическим и направлено на сохранение функции пораженных органов и улучшение качества жизни пациента [46, 48, 53, 55, 56].
Одной из приоритетных задач является купирование болевого синдрома, что требует комплексного подхода под наблюдением алголога или невролога. При наличии тревожности и депрессии показано обязательное направление к психиатру или психотерапевту для оказания соответствующей помощи.
Важнейший принцип хирургического лечения заключается в том, что выявление опухоли не служит прямым показанием к вмешательству. Решение об операции принимается индивидуально после тщательной оценки соотношения рисков и пользы для пациента. Хирургическое вмешательство показано только при наличии осложнений:
- для шванном – при некупируемой боли или неврологическом дефиците,
- для вестибулярных шванном (при NF2-шванноматозе) – при риске сдавления ствола мозга, потере слуха или нарушении функции лицевого нерва,
- для менингиом – по тем же принципам, что и для спорадических менингиом (например, при симптомном росте).
В медикаментозной терапии NF2-шванноматоза для лечения прогрессирующих вестибулярных шванном с целью уменьшения размера опухоли и стабилизации слуха применяется таргетный препарат бевацизумаб [57, 58]. Стандартная схема лечения на сегодняшний день не разработана; один из возможных режимов предполагает введение 5-7,5 мг/кг каждые 2-3 недели в течение не менее 6 месяцев с последующим переходом на поддерживающую терапию (2,5-5 мг/кг каждые 4 недели). Имеются данные о потенциальной эффективности бевацизумаба и при прогрессирующих кистозных эпендимомах.
Бессимптомные опухоли (например, медленно растущие интрамедуллярные эпендимомы или менингиомы) часто не требуют активного вмешательства и подлежат только регулярному клиническому и инструментальному мониторингу.
Список литературы
- Neurofibromatosis - StatPearls - NCBI Bookshelf [Электронный ресурс]. URL: https://www.ncbi.nlm.nih.gov/books/NBK459329/ (дата обращения: 28.08.2025).
- Plotkin S.R. и др. Updated diagnostic criteria and nomenclature for neurofibromatosis type 2 and schwannomatosis: An international consensus recommendation // Genetics in Medicine. Elsevier B.V., 2022. Т. 24, № 9. С. 1967–1977.
- Yoshida Y. Neurofibromatosis 1 (von Recklinghausen Disease) // Keio J Med. The Keio Journal of Medicine, 2025. Т. 74, № 1. С. 37–41.
- Stephens K. и др. Preferential mutation of the neurofibromatosis type 1 gene in paternally derived chromosomes // Hum Genet. Springer-Verlag, 1992. Т. 88, № 3. С. 279–282.
- Sobjanek M. и др. Segmental neurofibromatosis // Advances in Dermatology and Allergology/Postȩpy Dermatologii i Alergologii. Termedia Publishing House Ltd., 2014. Т. 31, № 6. С. 410.
- Lee T.S.J. и др. Incidence and prevalence of neurofibromatosis type 1 and 2: a systematic review and meta-analysis // Orphanet J Rare Dis. BioMed Central Ltd, 2023. Т. 18, № 1. С. 1–8.
- Ruggieri M., Huson S.M. The clinical and diagnostic implications of mosaicism in the neurofibromatoses // Neurology. Lippincott Williams and Wilkins, 2001. Т. 56, № 11. С. 1433–1443.
- Lugovskaya A.Y. и др. Нейрофиброматоз 1 типа в сочетании с феохромоцитомой: описание клинического случая с кратким обзором литературы // Problems of Endocrinology. Endocrinology Research Centre, 2023. Т. 70, № 2. С. 53.
- Rasmussen S.A., Yang Q., Friedman J.M. Mortality in neurofibromatosis 1: An analysis using U.S. death certificates // Am J Hum Genet. Elsevier, 2001. Т. 68, № 5. С. 1110–1118.
- Hirbe A.C. и др. Contemporary Approach to Neurofibromatosis Type 1–Associated Malignant Peripheral Nerve Sheath Tumors // American Society of Clinical Oncology Educational Book. Wolters Kluwer Health, 2024. Т. 44, № 3.
- Webster Carrion A., Shah A.C., Kotch C. Neurofibromatosis type 1-associated gliomas and other tumors: A new pathway forward? // Pediatric Hematology Oncology Journal. Elsevier, 2023. Т. 8, № 2. С. 129–135.
- de Traux de Wardin H. и др. NF1 -Driven Rhabdomyosarcoma Phenotypes: A Comparative Clinical and Molecular Study of NF1 -Mutant Rhabdomyosarcoma and NF1 -Associated Malignant Triton Tumor // JCO Precis Oncol. American Society of Clinical Oncology (ASCO), 2024. № 8.
- Spurr A. и др. An unusual case of pediatric embryonal rhabdomyosarcoma with subsequent diagnosis of neurofibromatosis type 1 // Pediatr Dermatol. John Wiley & Sons, Ltd, 2022. Т. 39, № 4. С. 664–666.
- Yukina M.Yu. и др. Атипичное и типичное течение нейрофиброматоза 1 типа в сочетании с феохромоцитомой // Эндокринная хирургия. Endocrinology Research Centre, 2022. Т. 15, № 3. С. 30–40.
- Wilson B.N. и др. Neurofibromatosis type 1: New developments in genetics and treatment // J Am Acad Dermatol. Mosby, 2021. Т. 84, № 6. С. 1667–1676.
- Legius E., Brems H. Genetic basis of neurofibromatosis type 1 and related conditions, including mosaicism // Child’s Nervous System. Springer Science and Business Media Deutschland GmbH, 2020. Т. 36, № 10. С. 2285–2295.
- Hsiao M.C. и др. Decoding NF1 Intragenic Copy-Number Variations // Am J Hum Genet. Cell Press, 2015. Т. 97, № 2. С. 238–249.
- Peduto C. и др. Neurofibromatosis Type 1: Pediatric Aspects and Review of Genotype–Phenotype Correlations // Cancers 2023, Vol. 15, Page 1217. Multidisciplinary Digital Publishing Institute, 2023. Т. 15, № 4. С. 1217.
- Scala M. и др. Genotype-phenotype correlations in neurofibromatosis type 1: A single-center cohort study // Cancers (Basel). MDPI AG, 2021. Т. 13, № 8. С. 1879.
- Sharif S. и др. A molecular analysis of individuals with neurofibromatosis type 1 (NF1) and optic pathway gliomas (OPGs), and an assessment of genotype–phenotype correlations // J Med Genet. BMJ Publishing Group Ltd, 2011. Т. 48, № 4. С. 256–260.
- Mo J. и др. Neurofibromin and suppression of tumorigenesis: beyond the GAP // Oncogene 2021 41:9. Nature Publishing Group, 2022. Т. 41, № 9. С. 1235–1251.
- Jiang C., McKay R.M., Le L.Q. Tumorigenesis in neurofibromatosis type 1: role of the microenvironment // Oncogene 2021 40:39. Nature Publishing Group, 2021. Т. 40, № 39. С. 5781–5787.
- Legius E. и др. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: an international consensus recommendation // Genetics in Medicine. Elsevier B.V., 2021. Т. 23, № 8. С. 1506–1513.
- Happle R. Superimposed Mosaicism in Neurocutaneous Syndromes // Neurocutaneous Disorders: A Clinical, Diagnostic and Therapeutic Approach: Third Edition. Springer, Cham, 2022. С. 17–24.
- Bruce R Korf, Mina Lobbous, Laura K Metrock. Neurofibromatosis type 1 (NF1): Pathogenesis, clinical features, and diagnosis - UpToDate [Электронный ресурс]. URL: https://www.uptodate.com/contents/neurofibromatosis-type-1-nf1-pathogenesis-clinical-features-and-diagnosis (дата обращения: 28.07.2025).
- Miettinen M.M. и др. Histopathologic evaluation of atypical neurofibromatous tumors and their transformation into malignant peripheral nerve sheath tumor in patients with neurofibromatosis 1—a consensus overview // Hum Pathol. W.B. Saunders, 2017. Т. 67. С. 1–10.
- PANDE A. и др. A Case of Atypical Neurofibromatous Neoplasm of Uncertain Biologic Potential: Unravelling the Enigma // JOURNAL OF CLINICAL AND DIAGNOSTIC RESEARCH. JCDR Research and Publications, 2025. Т. 19, № 2. С. 1.
- Rhodes S.D. и др. Cdkn2a (Arf) loss drives NF1-associated atypical neurofibroma and malignant transformation // Hum Mol Genet. Oxford University Press, 2019. Т. 28, № 16. С. 2752–2762.
- Higham C.S. и др. The characteristics of 76 atypical neurofibromas as precursors to neurofibromatosis 1 associated malignant peripheral nerve sheath tumors // Neuro Oncol. Oxford University Press, 2018. Т. 20, № 6. С. 818–825.
- Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference // Arch Neurol. 1988. Т. 45, № 5. С. 575.
- Neurofibromatosis type 1 (NF1): Management and prognosis - UpToDate [Электронный ресурс]. URL: https://www.uptodate.com/contents/neurofibromatosis-type-1-nf1-management-and-prognosis (дата обращения: 21.08.2025).
- Мустафин Р.Н. Клинические маски нейрофиброматоза 1-го типа // Архивъ внутренней медицины. SINAPS LLC, 2022. Т. 12, № 2. С. 93–103.
- Mir A. и др. Constitutional Mismatch Repair Deficiency Syndromes, a Neurofibromatosis 1 Mimicker That Hinders Timely Management // J Pediatr Hematol Oncol. Lippincott Williams and Wilkins, 2023. Т. 45, № 5. С. E613–E620.
- Denayer E., Legius E., Brems H. Legius Syndrome // The RASopathies: Genetic Syndromes of the RAS/MAPK Pathway. Springer, Cham, 2024. С. 63–77.
- Tamura R. Current Understanding of Neurofibromatosis Type 1, 2, and Schwannomatosis // International Journal of Molecular Sciences 2021, Vol. 22, Page 5850. Multidisciplinary Digital Publishing Institute, 2021. Т. 22, № 11. С. 5850.
- Collins-Sawaragi Y.C. и др. Location, symptoms, and management of plexiform neurofibromas in 127 children with neurofibromatosis 1, attending the National Complex Neurofibromatosis 1 service, 2018–2019 // Am J Med Genet A. John Wiley & Sons, Ltd, 2022. Т. 188, № 6. С. 1723–1727.
- Fisher M.J. и др. Management of neurofibromatosis type 1-associated plexiform neurofibromas // Neuro Oncol. Oxford Academic, 2022. Т. 24, № 11. С. 1827–1844.
- Mukhopadhyay S., Maitra A., Choudhury S. Selumetinib: the first ever approved drug for neurofibromatosis-1 related inoperable plexiform neurofibroma // Curr Med Res Opin. Taylor & Francis, 2021. Т. 37, № 5. С. 789–794.
- Imataka G. и др. Neurofibromatosis Type 1 and MEK Inhibition: A Comprehensive Review with Focus on Selumetinib Therapy // Journal of Clinical Medicine 2025, Vol. 14, Page 5071. Multidisciplinary Digital Publishing Institute, 2025. Т. 14, № 14. С. 5071.
- De Blank P.M.K. и др. MEK inhibitors for neurofibromatosis type 1 manifestations: Clinical evidence and consensus // Neuro Oncol. Oxford Academic, 2022. Т. 24, № 11. С. 1845–1856.
- Staedtke V. и др. Gene-targeted therapy for neurofibromatosis and schwannomatosis: The path to clinical trials // Clinical Trials. SAGE Publications Ltd, 2024. Т. 21, № 1. С. 51–66.
- Chong S. Lifelong Management of Neurofibromatosis 1 Patients // J Korean Neurosurg Soc. Korean Neurosurgical Society, 2025. Т. 68, № 3. С. 261–271.
- Brekelmans C. и др. Neurofibromatosis type 1-related pseudarthrosis: Beyond the pseudarthrosis site // Hum Mutat. John Wiley and Sons Inc., 2019. Т. 40, № 10. С. 1760–1767.
- Макашова Е.С. и др. Новая классификация и подходы к терапии шванноматозов // Журнал «Вопросы нейрохирургии» имени Н.Н. Бурденко. Media Sphera Publishing Group, 2023. Т. 87, № 5. С. 104–109.
- Planet M., Kalamarides M., Peyre M. Schwannomatosis: a Realm Reborn: year one // Curr Opin Oncol. Lippincott Williams and Wilkins, 2023. Т. 35, № 6. С. 550–557.
- Dhamija R. и др. LZTR1- and SMARCB1-Related Schwannomatosis // GeneReviews®. University of Washington, Seattle, 2024.
- Tsuchiya T. и др. Current molecular understanding of central nervous system schwannomas // Acta Neuropathologica Communications . BioMed Central Ltd, 2025. Т. 13, № 1.
- Evans D.G. NF2-Related Schwannomatosis // GeneReviews®. University of Washington, Seattle, 2023.
- Paganini I. и др. Expanding the mutational spectrum of LZTR1 in schwannomatosis // European Journal of Human Genetics. Nature Publishing Group, 2015. Т. 23, № 7. С. 963–968.
- Bigenzahn J.W. и др. LZTR1 is a regulator of RAS ubiquitination and signaling // Science (1979). American Association for the Advancement of Science, 2018. Т. 362, № 6419. С. 1171–1177.
- Kehrer-Sawatzki H., Cooper D.N. SMARCB1-related schwannomatosis and other SMARCB1-associated phenotypes: clinical spectrum and molecular pathogenesis // Fam Cancer. Springer Science and Business Media B.V., 2025. Т. 24, № 3. С. 64.
- NF2-related schwannomatosis (NF2-SWN; formerly neurofibromatosis type 2) - UpToDate [Электронный ресурс]. URL: https://www.uptodate.com/contents/nf2-related-schwannomatosis-nf2-swn-formerly-neurofibromatosis-type-2 (дата обращения: 28.08.2025).
- Schwannomatoses related to genetic variants other than NF2 - UpToDate [Электронный ресурс]. URL: https://www.uptodate.com/contents/schwannomatoses-related-to-genetic-variants-other-than-nf2?topicRef=5206&source=see_link (дата обращения: 28.08.2025).
- Perrino M.R. и др. Update on Cancer and Central Nervous System Tumor Surveillance in Pediatric NF2-, SMARCB1-, and LZTR1-Related Schwannomatosis // Clinical Cancer Research. American Association for Cancer Research Inc., 2025. Т. 31, № 8. С. 1400–1406.
- Рында А.Ю. и др. Комплексное лечение пациента с нейрофиброматозом 2 типа. // Журнал неврологии и психиатрии им. С.С. Корсакова. Media Sphera Publishing Group, 2020. Т. 120, № 8. С. 98–104.
- Schwannomatoses related to genetic variants other than NF2 - UpToDate [Электронный ресурс]. URL: https://www.uptodate.com/contents/schwannomatoses-related-to-genetic-variants-other-than-nf2?topicRef=5206&source=see_link (дата обращения: 21.08.2025).
- Douwes J.P.J. и др. Bevacizumab Treatment for Patients with NF2-Related Schwannomatosis: A Single Center Experience // Cancers 2024, Vol. 16, Page 1479. Multidisciplinary Digital Publishing Institute, 2024. Т. 16, № 8. С. 1479.
- Chiranth S. и др. A systematic review of targeted therapy for vestibular schwannoma in patients with NF2-related schwannomatosis // Neurooncol Adv. Oxford Academic, 2023. Т. 5, № 1.
Чтобы проверить свои знания материала, пройдите квиз. Кликните на картинку, чтобы начать.

Автор статьи

Статьи по теме Онкология
Болезни в статье:
- C47 Злокачественное новообразование периферических нервов и вегетативной нервной системы
- H46-H48 Болезни зрительного нерва и зрительных путей
- C49 Злокачественное новообразование других типов соединительной и мягких тканей
- C74 Злокачественное новообразование надпочечника
- C71 Злокачественное новообразование головного мозга
- C81-C96 Злокачественные новообразования лимфоидной, кроветворной и родственных им тканей
- C50 Злокачественные новообразования молочной железы
- C15-C26 Злокачественные новообразования органов пищеварения
- Q77 Остеохондродисплазия с дефектами роста трубчатых костей и позвоночника
- M84.1 Несрастание перелома [псевдоартроз]
- E34.3 Низкорослость [карликовость], не классифицированная в других рубриках
- M41 Сколиоз
- M85 Другие нарушения плотности и структуры кости
- M81 Остеопороз без патологического перелома
- F84 Общие расстройства психологического развития
- F90.0 Нарушение активности и внимания
- G40-G47 Эпизодические и пароксизмальные расстройства
- Q25.6 Стеноз легочной артерии
- Q21 Врожденные аномалии [пороки развития] сердечной перегородки
- Q25.1 Коарктация аорты
- Q21.3 Тетрада Фалло
- I34.1 Пролапс [пролабирование] митрального клапана
- I42 Кардиомиопатия
- I10-I15 Болезни, характеризующиеся повышенным кровяным давлением
- K59.0 Запор
- K58 Синдром раздраженного кишечника
- K30 Диспепсия
Фармгруппы в статье:
Оцените статью:
Полужирным шрифтом выделены лекарства, входящие в справочники текущего года. Рядом с названием препарата может быть указан ежегодный уровень индекса информационного спроса (показатель, который отражает степень интереса потребителей к информации о лекарстве).
нейрофиброматоз
нейрофиброматоз,События
Реклама: ООО «Конгресслайн», ИНН 7708369172
Реклама
