C90 Множественная миелома и злокачественные плазмоклеточные новообразования, МКБ-10
Отредактировано: 02.04.2024
к.м.н. Вотякова О.М.
ГУ РОНЦ им. Н.Н. Блохина РАМН
Заболеваемость. Факторы риска. Патоморфология. Патогенез. Клиническая картина. Диагностика. Дифференциальный диагноз. Клиническая классификация. Прогноз и прогностические факторы. Лечени
Множественная миелома (миеломная болезнь, болезнь Рустицкого — Калера) — это злокачественное лимфопролиферативное заболевание, морфологическим субстратом которого являются плазматические клетки, продуцирующие моноклональный иммуноглобулин (парапротеин, М-градиент). Моноклональная продукция характеризуется синтезом структурно полноценных молекул иммуноглобулинов, их фрагментов или сочетанием тех и других.
Множественная миелома, а также солитарные плазмоцитомы, лимфоплазмоцитарная лимфома/макроглобулинемия Вальденстрема (иммуноцитома) и болезни тяжелых цепей входят в группу злокачественных опухолей под общим названием “парапротеинемические гемобластозы”. В соответствии с Европейско-американской классификацией (REAL) и классификацией ВОЗ (WHO) множественная миелома относится к периферическим В-клеточным лимфоидным опухолям.
Заболеваемость
Множественная миелома составляет 1% от всех онкологических заболеваний и немногим более 10% среди всех гемобластозов. Заболевание встречается у людей всех рас и на всех континентах. Самая низкая заболеваемость наблюдается в Китае: 1,0 на 100 тыс. населения. В США этот показатель среди мужчин и женщин афроамериканцев составляет 9,9 и 6,7 на 100 тыс. населения соответственно. У жителей США белой расы множественная миелома встречается реже: у мужчин — 4,3 и у женщин — 3,0 на 100 тыс. населения. Заболеваемость множественной миеломой в Европе в 2000 г. составила 1,31; а в России — 1,24 на 100 тыс. населения.
Болеют множественной миеломой преимущественно в возрасте старше 40 лет, средний возраст больных составляет 69 лет.
Факторы риска
Причины развития множественной миеломы у человека остаются неясными. В качестве возможных этиологических факторов обсуждается значение ионизирующей радиации, генетической предрасположенности, длительной антигенной стимуляции, воздействия токсических веществ.
Выделение из костного мозга больных множественной миеломой вируса герпеса человека VIII типа (HHV–8, известного как вирус саркомы Капоши), а также его участие в развитии ряда лимфопролиферативных заболеваний, позволили предположить, что HHV–8 играет важную роль в патогенезе множественной миеломы. Однако это предположение пока не получило убедительных доказательств.
Патоморфология
Морфологическим субстратом множественной миеломы и солитарной плазмоцитомы чаще всего являются плазмоциты, однако могут присутствовать и плазмобласты. Как среди плазмоцитов, так и плазмобластов встречаются многоядерные формы. Преобладание в опухолевой ткани плазмобластов, проплазмоцитов, а также атипичных клеточных форм является прогностически неблагоприятным морфологическим признаком. В экстрамедуллярных очагах в ткани плазмоцитомы обычно много синусоид, придающих опухоли ячеистое строение.
Патогенез
До настоящего времени остается неясным вопрос о том, на каком этапе дифференцировки происходит трансформация нормальной лимфоидной В-клетки в опухолевую с дальнейшим неконтролируемым клональным ростом. Исследования последних лет позволяют лишь предположить, что предшественники миеломных клеток образуются в периферических лимфоидных органах.
Опухолевые клетки при множественной миеломе проявляют важные свойства нормальных долгоживущих плазматических клеток — потомков В-лимфоцитов, прошедших этап стимуляции антигеном, соматических гипермутаций и изотипического переключения синтеза антител. Предполагается, что во время созревания В-клетки происходят ошибки, которые приводят к хромосомным транслокациям с вовлечением генов иммуноглобулинов, в частности локуса тяжелой цепи (IgH) на 14 хромосоме — область 14q32. По всей вероятности предполагаемыми предшественниками миеломных клеток являются В-лимфоциты, которые уже прошли фолликулярный центр лимфатических узлов. Сказанное подтверждается тем, что при множественной миеломе доминирует фенотип опухолевых клеток после изотипического переключения синтеза иммуноглобулинов, т.е. подвергшихся соматическим гипермутациям.
Важную роль в процессе роста опухоли играют цитокины, секретируемые миеломными клетками и стромальными элементами костного мозга. Основным фактором роста миеломных клеток является интерлейкин–6 (ИЛ–6), секретируемый в основном стромальным микроокружением. В миеломных клетках ИЛ–6 активирует трансдуцерную молекулу gp130, пути внутриклеточной передачи сигнала JAK/STAT и другие. В результате такой активации происходит стимуляция пролиферативной активности миеломных клеток при одновременном резком снижении их способности к апоптозу.
Фактор некроза опухоли альфа (tumor necrosis factor — ТНФ альфа) является другим цитокином, играющим важную роль во взаимодействии между опухолевыми миеломными клетками и клетками стромы костного мозга. Фактор некроза опухоли альфа регулирует синтез молекул адгезии на миеломных (LFA–1, VLA–4) и стромальных клетках (ICAM–1, VCAM–1). В результате происходит усиление адгезии (прилипания) миеломных клеток к стромальным клеткам костного мозга, что в свою очередь приводит к увеличению секреции ИЛ–6 костно-мозговым микроокружением.
Стимулирующее влияние на пролиферацию опухолевых плазматических клеток при множественной миеломе могут оказывать и другие цитокины: ГМ-КСФ (GM-CSF), ИЛ–1, ИЛ–3, ИЛ–5.
Известно, что миеломные клетки продуцируют ИЛ–1 бета, ТНФ-бета, моноцитарно-макрофагальный колониестимулирующий фактор (М-КСФ). Они стимулируют стромальные клетки костного мозга, а также остеокласты, ответственные за резорбцию костей, т.е. играют важную роль в патофизиологии остеодеструктивного процесса.
Некоторые цитокины ингибируют рост миеломных клеток. Наиболее мощным ингибитором роста плазматических клеток in vitro является гамма-интерферон. ИЛ–4 также замедляет пролиферацию миеломных клеток in vivo. Выраженной антипролиферативной активностью обладает интерферон альфа.
Изучение кариотипа миеломных клеток затруднено из-за их низкой пролиферативной активности. Аномалии кариотипа при использовании стандартных цитогенетических методов выявляются у 30–50% больных. Применение метода флуоресцентной гибридизации in situ (FISH) позволяет проводить цитогенетические исследования в неделящихся клетках. Методом FISH хромосомные аномалии обнаруживаются у 89–96% больных множественной миеломой.
Данные кариологических исследований свидетельствуют о хромосомной нестабильности, проявляющейся количественными и структурными изменениями хромосом. Наиболее характерными количественными аномалиями кариотипа при множественной миеломе являются: моносомия 13, трисомия 3, 5, 7, 9, 15 и 19. Делеция короткого плеча 17 хромосомы (17р13) с утратой или мутацией гена супрессора опухолевого роста р53 выявляется у 30–35% первичных больных множественной миеломой и гораздо чаще на поздних стадиях развития болезни, а также при агрессивно протекающей миеломе. Структурные аберрации наиболее часто вовлекают хромосомы: 1 (оба плеча), 6q, 11q и 14q32 (IgH локус).
Специфические изменения кариотипа при множественной миеломе чаще всего характеризуются наличием транслокаций с участием 14q32: t(11;14)(q13;q32), t(4;14)(p16;q32), t(14;16)(q32;q23). Транслокации с вовлечением участка 14q32 захватывают район гена, контролирующего изотипическое переключение синтеза тяжелых цепей Ig. Исследования методом FISH показали, что транслокации c вовлечением области 14q32 обнаруживаются приблизительно у 50% больных с моноклональной гаммапатией неясного генеза (MGUS — monoclonal gammopathies of undetermined significance), у 75% — миеломой и более чем у 80% — плазмоклеточным лейкозом. Предполагается, что перестройки 14q32 имеют значение в патогенезе множественной миеломы, являясь одной из причин злокачественной трансформации, а делеция или мутация гена р53, скорее, отвечает за опухолевую прогрессию.
В последние годы установлено, что отдельные хромосомные аномалии имеют важное прогностическое значение при множественной миеломе и определяют ответ на лечение и выживаемость. Так, при выявлении делеции длинного плеча хромосомы 13 или моносомии 13, в большинстве случаев отмечается рефрактерность как к стандартной, так и к высокодозной химиотерапии (ВДХ). Чрезвычайно агрессивное течение болезни и короткая выживаемость наблюдаются у больных с транслокациями t(4;14)(p16,3;q32) и с t(14;16)(q32;q23). Прогноз множественной миеломы хуже при гиподиплоидном кариотипе в сравнении с гипердиплоидным. У больных с делецией короткого плеча 17 хромосомы с утратой или мутацией гена супрессора опухолевого роста р53 также отмечается короткая продолжительность жизни.
Фенотип злокачественных плазматических клеток имеет 2 особенности. Первая заключается в том, что в процессе трансформации В-лимфоцитов в плазматические клетки происходит утрата большинства В-линейных маркеров. Второй особенностью является приобретение в процессе трансформации большого количества адгезивных структур.
Наиболее важными иммунологическими маркерами плазматических клеток являются CD138 (синдекан–1) и CD38. Кроме того, для плазматических клеток, как нормальных, так и опухолевых, характерна экспрессия CD54 (ICAM–1), CD44 (HCAM), CD24 и отсутствие или низкий уровень экспрессии CD45. Основным иммунологическим маркером, позволяющим дифференцировать опухолевые и неопухолевые плазматические клетки, является антиген CD19. Нормальные плазматические клетки обычно сохраняют способность к экспрессии CD19, в то время как для миеломных клеток экспрессия CD19 не характерна. Кроме того, на поверхности миеломных клеток часто выявляются СD58 (LFA–1) и CD56 (NCAM). Эти маркеры обычно на нормальных плазматических клетках не обнаруживаются.
Предполагается, что CD56 включается в процессы адгезии и имеет отношение к остеолизу. Отсутствие экспрессии CD56 ассоциируется с более агрессивным течением множественной миеломы и тенденцией к увеличению плазматических клеток в периферической крови. При плазмоклеточном лейкозе CD56 на опухолевых клетках крови и костного мозга не обнаруживается.
Выраженная экспрессия CD28 выявляется при высокой пролиферативной активности миеломных клеток обычно при прогрессировании и рецидивах болезни.
В последние годы большое значение в патогенезе множественной миеломы придается опухолевому ангиогенезу. Миеломные клетки синтезируют факторы роста эндотелия сосудов (VEGF — vascular endothelial growh factor) и металлопротеиназы (МР), которые путем взаимодействия с рецепторами на клетках стромы, стимулируют секрецию ИЛ–6 и ТНФ-альфа. С одной стороны, VEGF, МР усиливают процесс неоваскуляризации сосудов опухоли, а с другой стороны, способствуют пролиферации миеломных клеток.
Клиническая картина
Клинические проявления заболевания при множественной миеломе обусловлены плазмоклеточной инфильтрацией костного мозга, костей, внекостномозговым распространением опухоли и секрецией моноклонального иммуноглобулина.
Одно из основных проявлений болезни — поражение костей. У 70% больных отмечаются боли в костях. Остеодеструкции чаще развиваются в плоских костях (череп, таз, грудина, ребра), позвонках, а также в проксимальных отделах бедренных и плечевых костей (рис. 1). Поражение скелета может сопровождаться костными деформациями. В терминальной стадии заболевания и при агрессивно текущей миеломе возможно прорастание опухоли в мягкие ткани. Классический симптом — спонтанные переломы. Переломы позвонков возникают у половины больных множественной миеломой, а переломы других костей — в 30% случаев.
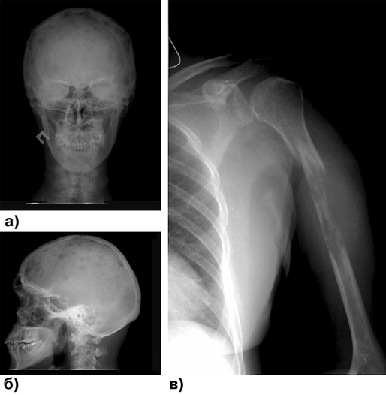
Рисунок 1. Поражения черепа (а, б) и плечевой кости (в) у больной множественной миеломой
Рентгенологически поражение костной ткани у большинства больных множественной миеломой выявляется в виде генерализованного остеопороза, единичных или множественных очагов остеолиза, патологических переломов.
Повышение содержания кальция в сыворотке крови наблюдается у 20–30% больных множественной миеломой. Оно обусловлено усиленной резорбцией костей и наиболее выражено у больных с массивными остеолитическими поражениями. Гиперкальциемия клинически проявляется потерей аппетита, тошнотой, рвотой, запорами, полиурией, гипотонией, мышечной слабостью, нарушениями сердечного ритма. У части больных повышение уровня кальция может быть бессимптомным. Одним из тяжелых проявлений гиперкальциемии является почечная недостаточность, которая при отсутствии своевременного лечения может привести к развитию комы и смерти больного.
При множественной миеломе довольно часто наблюдается поражение почек. Это характеризуется протеинурией, постепенным снижением концентрационной функции почек, развитием хронической или острой почечной недостаточности. Почечная недостаточность развивается более чем у 50% больных множественной миеломой и среди причин смерти занимает 2 место после инфекций.
Патогенез поражения почек при множественной миеломе сложен. Основным фактором повреждения почек является избыточная секреция легких цепей иммуноглобулинов (белка Бенс-Джонса). Секреция белка Бенс-Джонса может стать причиной миеломной нефропатии, амилоидоза и болезни отложения легких цепей иммуноглобулинов, почечно-канальцевой дисфункции (приобретенный синдром Фанкони).
Миеломная нефропатия — наиболее частая причина развития почечной недостаточности. Реабсорбция легких цепей иммуноглобулинов приводит как к повреждению эпителия канальцев и нарушению их функции, так и к образованию цилиндров внутри канальцев с последующей их обструкцией. Специфических клинических симптомов миеломной нефропатии нет. У большинства больных в моче выявляются моноклональные легкие цепи иммуноглобулинов и относительно небольшое количество белка. Альбуминурия более чем 1 г/сут обычно связана с амилоидозом или болезнью отложения легких цепей иммуноглобулинов.
При болезни отложения легких цепей иммуноглобулинов и амилоидозе легкие цепи иммуноглобулинов и их фрагменты образуют депозиты в почках и других органах. Амилоидные фибриллы формируются, главным образом лямбда-цепями иммуноглобулинов, выявляются при окраске Конго красным и при электронной микроскопии. При болезни отложения легких цепей иммуноглобулинов депозиты образуются в основном каппа-цепями иммуноглобулинов, имеют аморфный не фибриллярный состав и не выявляются гистологическими методами, принятыми для диагностики амилоидоза. Клинически поражение почек при амилоидозе и болезни отложения легких цепей иммуноглобулинов проявляется протеинурией, нефротическим синдромом, реже почечной недостаточностью.
При синдроме Фанкони, развивающемся иногда у больных множественной миеломой, токсическое действие легких цепей иммуноглобулинов ограничивается нарушением функции почечных канальцев. Этот синдром проявляется почечной глюкозурией, аминоацидурией, гипофосфатемией, хроническим ацидозом, гипоурикемией и гипокалиемией. Классические признаки нефротического синдрома: отеки, гиперхолестеринемия, обычно отсутствуют. Синдром Фанкони обычно сопровождается остеомаляцией (размягчением костей).
Кроме протеинурии Бенс-Джонса развитию почечной недостаточности при множественной миеломе способствуют и другие факторы: гиперурикемия при распаде большой массы опухоли, дегидратация, гиперкальциемия, гемодинамические нарушения, связанные с анемией или гипервязкостью плазмы. Инфекция мочевыводящих путей редко является самостоятельной причиной развития почечной недостаточности, но присоединение ее резко ухудшает прогноз.
При нарушении функции почек у больных множественной миеломой назначение любого лекарственного препарата должно быть оценено с учетом возможного нефротоксического действия. Использование целого ряда лекарственных средств — аминогликозидов, амфотерицина В, НПВС, ингибиторов ангиотензинконвертазы, ацикловира, может стать причиной развития почечной недостаточности.
При установлении диагноза множественной миеломы следует избегать введения рентгеноконтрастных препаратов, т.к. в их присутствии может произойти выпадение белка Бенс-Джонса в почечных канальцах с последующей необратимой анурией, особенно у обезвоженных больных.
Амилоидоз развивается у 10–15% больных множественной миеломой. Амилоидозом при множественной миеломе поражаются главным образом органы, богатые коллагеном: адвентиций сосудов, сердечная мышца, язык, дерма, нервы, сухожилия, суставы. Наиболее частыми проявлениями являются слабость, утомляемость, снижение массы тела. Амилоидоз печени и селезенки, как правило, протекает бессимптомно, но может сопровождаться гепатоспленомегалией.
Развитие амилоидоза у больных множественной миеломой может быть заподозрено при макроглоссии, геморрагическом синдроме, синдроме карпального канала, сердечной недостаточности, нефротическом синдроме, разнообразных дерматозах, артралгиях с деформациями суставов, диспептических расстройствах, дистрофии роговицы. Почечная недостаточность относится к поздним проявлениям амилоидоза. Для установления амилоидоза необходимо гистологическое исследование материала, полученного при биопсии слизистой оболочки прямой кишки, либо изучение аспирата подкожного жира в области передней брюшной стенки.
При болезни отложения легких цепей иммуноглобулинов депозиты легких каппа-цепей образуются не только в почках, но и в других органах, чаще в сердце, печени, нервной системе, ЖКТ. Заболевание протекает без каких-либо специфических симптомов, поэтому редко диагностируется клинически.
Повышенный уровень сывороточных иммуноглобулинов, обусловленный продукцией моноклонального белка, может привести к увеличению вязкости крови, которая при достижении определенного уровня становится причиной разнообразных осложнений. Синдром гипервязкости чаще развивается у больных множественной миеломой при высоком уровне моноклональных IgA или IgG3, склонных к полимеризации. Клиническая картина этого синдрома характеризуется кровоточивостью, ретинопатией с характерными пламенеющими кровоизлияниями, расширением вен сетчатки, парестезиями, синдромом Рейно. Нарушение микроциркуляции в сосудах головного мозга может стать причиной неврологических расстройств, вплоть до парапротеинемической комы.
Одним из частых клинических проявлений множественной миеломы является разнообразная неврологическая симптоматика. У 10–20% больных в начале или по ходу течения болезни может наблюдаться компрессия спинного мозга. Это осложнение может быть связано со сдавлением спинного мозга фрагментами разрушенного позвонка или экстрадурально расположенной опухолью. Клинически компрессия спинного мозга проявляется радикулярными болями, мышечной слабостью, парезами или параплегиями нижних конечностей, нарушением функции тазовых органов
Из других неврологических осложнений наиболее часто встречаются корешковые боли и периферическая нейропатия. Внутричерепные плазмоцитомы, специфическое поражение мягкой и паутинной оболочек, а также черепно-мозговых нервов встречаются редко. Клинически поражение оболочек мозга проявляется упорной головной болью.
Корешковые боли возникают, главным образом, в грудном или пояснично-крестцовом отделах позвоночника. Они могут быть вызваны компрессией нервов при поражении или разрушении позвонков, а также плазмоклеточной инфильтрацией корешков.
У 5–15% больных при множественной миеломе наблюдается периферическая нейропатия. Механизм развития этого осложнения не ясен. Нейропатия проявляется нарушением тактильной и болевой чувствительности, парестезиями, мышечной слабостью, онемением, болями. Проявления нейропатии обычно симметричны. При гистологическом исследовании выявляются дегенерация и демиелинизация аксонов, а в некоторых случаях отложения амилоида. В качестве возможных причин развития нейропатии обсуждается токсическое воздействие на аксоны парапротеина, иммунных комплексов, метаболических факторов, лекарственных препаратов (винкристин, цисплатин). Случаи остеосклеротической миеломы с полинейропатией, органомегалией, эндокринопатией и поражением кожи характеризуются как POEMS-синдром (P-polyneuropathy, O-organomegaly, E-endocrinopathy, M-M protein, S-skin).
У 5–13% больных множественной миеломой выявляется гепатомегалия или спленомегалия. В половине случаев увеличение органов связано со специфической плазмоклеточной пролиферацией. Поражение лимфатических узлов в развернутой стадии множественной миеломы встречается редко и составляет менее 0,5%. Специфическое поражение желудка может проявляться в виде инфильтративного процесса, язвы или плазмоцитомы. Опухолевые плазмоклеточные инфильтраты могут развиваться практически во всех органах, но редко проявляются клинически. Обычно их обнаруживают при аутопсии.
В силу ряда причин, прежде всего из-за подавления синтеза нормальных иммуноглобулинов, у больных множественной миеломой снижена способность противостоять инфекции. Частота инфекций при этой болезни в 15 раз выше, чем у здоровых людей. Инфекционные осложнения являются основной причиной смерти больных множественной миеломой. Наиболее часто развиваются пневмония и инфекции мочевыводящих путей. В ранней стадии болезни основными возбудителями инфекции являются S. pneumonia и H. influenzae. В поздних стадиях — инфекции чаще обусловлены грамотрицательными возбудителями и стафилококками.
К факторам риска развития инфекционных осложнений относятся: длительная нейтропения, установка центральных венозных катетеров, назначение высоких доз глюкокортикоидов. В последнее десятилетие из-за широкого внедрения в клиническую практику метода высокодозной химиотерапии (ВДХ), а также в связи с использованием глюкокортикоидов в высоких дозах отмечается увеличение частоты развития грибковых инфекций. Кроме того, увеличилась заболеваемость опоясывающим герпесом и пневмонией, вызываемой P. carini.
Лихорадка без видимой инфекционной причины, а также другие симптомы интоксикации (Б симптомы), при множественной миеломе наблюдаются чрезвычайно редко.
Кровоточивость встречается у 15% больных с множественной миеломой, чаще при IgA-миеломе. Геморрагический синдром обычно связан с нарушениями в тромбоцитарном звене гемостаза вследствие цитостатической тромбоцитопении. Кроме того, геморрагии могут быть обусловлены нарушением функции тромбоцитов за счет “обволакивания” их парапротеином. Кровоточивость может быть связана и с нарушениями прокоагулянтного звена гемостаза — ингибированием факторов свертывания крови антикоагулянтами, продуцируемыми плазматическими клетками.
У 60–70% больных множественной миеломой при установлении диагноза выявляется анемия, как правило, нормоцитарная, нормохромная. По мере прогрессирования болезни анемия развивается у всех больных миеломой. Развитие анемии обычно связано со снижением выработки эндогенного эритропоэтина. Определенную роль играют и другие факторы: опухолевая инфильтрация костного мозга, миелосупрессивное действие химиопрепаратов, токсическое влияние на костный мозг “уремических факторов” при почечной недостаточности, укорочение продолжительности жизни эритроцитов, дефицит железа.
Повышение СОЭ отмечается в 70% случаев и связано с присутствием парапротеина в сыворотке крови. Варианты болезни с низкой секрецией сывороточного парапротеина и миелома Бенс-Джонса протекают с нормальной СОЭ. Лейкопения и тромбоцитопения при множественной миеломе встречаются редко. Их развитие, как правило, свидетельствует либо о выраженной инфильтрации костного мозга миеломными клетками, либо о миелосупрессии после химиотерапии. Иногда отмечается нейтрофилез со сдвигом лейкоцитарной формулы влево. В крови может выявляться небольшое количество плазматических клеток. Высокий плазмоцитоз крови, как правило, является проявлением плазмоклеточного лейкоза. На начальных этапах заболевания иногда определяются тромбоцитоз и увеличение количества мегакариоцитов в миелограмме.
Гиперпротеинемия обусловлена повышенной продукцией парапротеина. Для выявления моноклонального иммуноглобулина необходимы иммунохимические исследования: электрофорез белков сыворотки крови (рис. 2) и мочи (рис. 3), иммуноэлектрофорез и иммунофиксация. Характерным симптомом множественной миеломы является снижение уровня нормальных иммуноглобулинов.
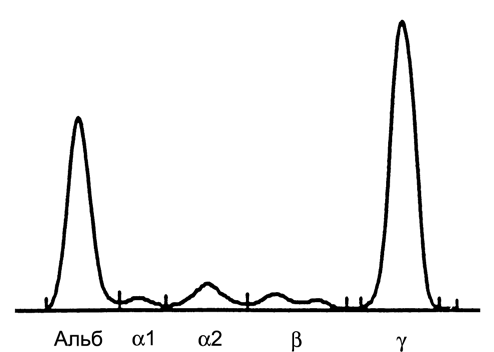
Рисунок 2. Электрофоретическое исследование белков сыворотки крови у больной множественной миеломой:
M-градиент в зоне γ2 образован парапротеином Gκ и составляет 51% от общего белка сыворотки крови или 55,5 г/дл
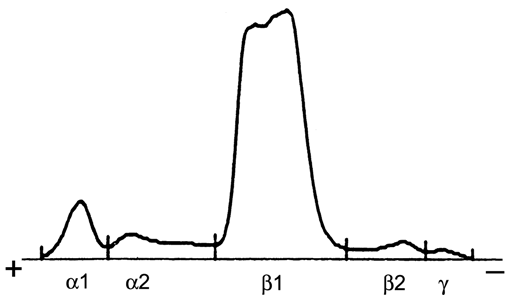
Рисунок 3. Электрофоретическое исследование мочи у больной множественной миеломой:
M-градиент в β-зоне образован белком Бенс-Джонса κ-типа и составляет 77,5% от суточного белка мочи или 4,88 г/л
При исследовании костного мозга у 90–96% больных отмечается плазмоклеточная инфильтрация. При очаговом характере опухолевого поражения костного мозга специфические изменения в миелограмме могут не обнаруживаться. В таких случаях показаны повторные пункции грудины и/или подвздошной кости, трепанобиопсия подвздошной кости или других пораженных участков скелета.
В зависимости от особенностей клинического течения и лабораторных признаков кроме типичной миеломы выделяются следующие редкие варианты заболевания: несекретирующая, тлеющая, вялотекущая миелома и плазмоклеточный лейкоз.
Несекретирующая миелома характеризуется отсутствием в сыворотке крови и моче М-градиента. Плазматические клетки при этом варианте болезни синтезируют, но не секретируют молекулы Ig. Использование метода иммунофлуоресценции с моноспецифическими сыворотками позволяет выявить в большинстве случаев в цитоплазме плазматических клеток моноклональный Ig. При несекретирующей миеломе редко развивается почечная недостаточность, снижение уровня нормальных Ig выражено незначительно.
Тлеющая (smouldering) миелома характеризуется бессимптомным течением. Общее состояние больных, как правило, не страдает. Костные поражения отсутствуют. Анемия, почечная недостаточность и гиперкальциемия не наблюдаются. При первичной диагностике этот вариант болезни трудно отличить от типичной множественной миеломы, так как уровень М-компонента обычно превышает 30 г/л, а количество плазматических клеток в костном мозге выше 10%.
Вялотекущая (индолентная) миелома. Этот вариант заболевания подобен тлеющей миеломе, с тем отличием, что рентгенологически могут выявляться единичные очаги поражения в костях (не более трех). Боли в костях и компрессионные переломы не наблюдаются. Анемия умеренная (уровень Нb не ниже 100 г/л).
Плазмоклеточный лейкоз развивается редко и составляет 2%. В периферической крови абсолютное количество плазматических клеток превышает 2х109 /л. В лейкоцитарной формуле число плазматических клеток выше 20%. Выделяют две формы плазмоклеточного лейкоза: первичный, выявляемый при установлении диагноза множественной миеломы, и вторичный, когда плазмоклеточная лейкемизация развивается в терминальной фазе болезни.
При плазмоклеточном лейкозе часто обнаруживаются экстрамедуллярные очаги опухолевого роста с поражением печени, селезенки, лимфоузлов, менингеальных оболочек. Заболевание характеризуется агрессивным течением и короткой продолжительностью жизни. Лечение плазмоклеточного лейкоза малоэффективно.
Приблизительно в 3–7% случаев наблюдается одиночное поражение костей или мягких тканей — солитарная плазмоцитома. При этой форме заболевания отсутствует поражение костного мозга, парапротеин в сыворотке крови и/или в моче обычно не определяется, либо его количество незначительно. Выделяют солитарную костную и внекостную (мягкотканную) плазмоцитомы. Солитарные очаги в костях чаще локализуются в позвонках, ребрах, черепе, ключицах, лопатках, а также бедренных костях и костях таза. Мягкотканная плазмоцитома чаще всего (до 80%) локализуется в верхних дыхательных путях: придаточных пазухах носа, носоглотке и ротоглотке. Плазмоцитома может обнаруживаться также в ЖКТ, ЦНС, щитовидной железе, паращитовидных железах, молочных железах, лимфатических узлах, мочевом пузыре, яичках и коже. Основным методом лечения солитарных плазмоцитом является лучевая терапия. Эффективность лечения выше у больных с поражением мягких тканей.
Диагностика
При диагностике множественной миеломы основными являются три критерия:
1) обнаружение более 10% плазматических клеток в миелограмме и (или) плазмоклеточной опухоли в биопсийном материале пораженной ткани;
2) выявление моноклонального Ig методом иммуноэлектрофореза (в сыворотке крови содержание IgG более 35 г/л или IgA более 20 г/л, в моче каппа— или лямбда-легкие цепи более 1 г/сут);
3) наличие остеолитических поражений скелета и/или диффузный остеопороз.
Диагноз множественной миеломы может считаться достоверным только при сочетании не менее двух из трех основных критериев, причем наличие плазмоклеточного опухолевого поражения обязательно.
Объем исследований, необходимый для установления диагноза множественной миеломы:
I.Обязательные исследования.
1. Рентгенография костей:
— череп, ключицы, лопатки, грудина, ребра, все отделы позвоночника, кости таза, проксимальные отделы плечевых и бедренных костей;
— другие отделы скелета при наличии клинических проявлений.
2. Биохимический анализ крови с определением уровня общего белка, альбумина, кальция, креатинина, мочевой кислоты, ЛДГ, щелочной фосфатазы, билирубина, трансаминаз.
3. Общий анализ крови с подсчетом лейкоцитарной формулы и числа тромбоцитов.
4. Исследование уровня иммуноглобулинов в сыворотке крови методом радиальной иммунодиффузии.
5. Электрофорез белков сыворотки крови с количественным определением уровня М-градиента (денситометрия).
6. Общий анализ мочи.
7. Исследование мочи по Зимницкому.
8. Исследование суточной мочи для определения потери белка с мочой.
9. Электрофорез белков концентрированной мочи с количественным определением уровня парапротеина.
10. Морфологическое исследование костного мозга, а в отсутствие его поражения — опухолевой ткани.
II. Дополнительные исследования.
1. Определение в сыворотке крови уровня бета2-микроглобулина, С реактивного белка.
2. Определение митотической активности плазматических клеток по индексу метки (LI) тритий тимидином или бромдезоксиуридином.
3. МРТ при подозрении на сдавление спинного мозга.
4. Цитогенетическое исследование плазматических клеток с использованием FISH-метода.
5. Иммунофенотипирование опухолевых плазматических клеток.
Дифференциальный диагноз
Установить диагноз множественной миеломы не всегда просто, т.к. моноклональный белок может выявляться в сыворотке крови и/или в моче при различных патологических состояниях. Это может иметь место при других В-клеточных лимфопролиферативных заболеваниях: неходжкинских лимфомах, хроническом лимфолейкозе. Наличие парапротеина может быть одним из проявлений амилоидоза. Множественные поражения костей, сопровождающиеся плазмоклеточной инфильтрацией костного мозга и парапротеинемией, встречаются при некоторых формах рака с метастазами в кости и костный мозг. М-градиент может выявляться при некоторых синдромах и состояниях, сопровождающихся повышенной продукцией антител: реконвалесценция после пневмонии, гепатит, паразитарные инфекции, цирроз печени, саркоидоз Бека, аллергические реакции, аутоиммунные заболевания.
В ряде случаев М-протеин выявляется у практически здоровых людей. В таких случаях речь может идти о моноклональной гаммапатии неясного генеза — МГНГ (MGUS). Эта гаммапатия выявляется у 5% здорового населения в возрасте от 22 до 65 лет. Частота МГНГ увеличивается с возрастом и достигает 10% у людей старше 80 лет. При этом отсутствуют клинические симптомы множественной миеломы. Литические костные поражения на рентгенограммах не обнаруживаются. В миелограмме при МГНГ число плазматических клеток не превышает 10%. Уровень сывороточного М-компонента не выше 35 г/л для IgG и 20 г/л для IgA, уровни других иммуноглобулинов в пределах нормы. В моче М-компонент менее 1 г/сут. Обычно пациенты с МГНГ это пожилые люди. У трети пациентов с МГНГ, в конечном счете, развивается множественная миелома или другие лимфопролиферативные заболевания.
Клиническая классификация
Миелома подразделяется на варианты по типу продуцируемого моноклонального иммуноглобулина. Частота распределения различных иммунохимических вариантов опухоли приблизительно соответствует концентрации различных классов нормальных иммуноглобулинов в сыворотке (табл. 1).
Таблица 1
Иммунохимические варианты множественной миеломы
| Вариант | PIg* сыворотки | PIg мочи (белок Бенс–Джонса) | Частота, % |
| G-миелома | Gκ | κ | 55–65 |
| Gλ | λ | ||
| Gκ или Gλ | Нет | ||
| А-миелома | Аκ | κ | 20–25 |
| Аλ | λ | ||
| Аκ или Аλ | Нет | ||
| D-миелома | Dκ | κ | 2–5 |
| Dλ | λ | ||
| Dκ или Dλ | Нет | ||
| E-миелома | Еκ | κ | Точно не установлена |
| Еλ | λ | ||
| Еκ или Еλ | Нет | ||
| Миелома Бенс–Джонса | Нет | κ | 12–20 |
| Нет | λ | ||
| Несекретирующая миелома | Нет | Нет | 1–4 |
| Диклональные миеломы | Разные соотношения двух и более PIg | κ или λ | 1–2 |
| М-миелома | Мκ | κ | 0,5 |
| Мλ | λ | ||
| Мκ или Мλ | Нет |
* PIg — моноклональный Ig (парапротеин)
Распределение миеломы по стадиям производится по клинико-лабораторным показателям, косвенно отображающим опухолевую массу (табл. 2).
Таблица 2
Стадии множественной миеломы
(Durie B.G.M., Salmon S.E., 1975)
| Стадии | Признаки | Масса миеломных клеток, 109/м2 |
| I | Все из перечисленных: гемоглобин более 100 г/л, нормальный уровень кальция сыворотки, рентгенологически-нормальная структура костей или солитарный очаг плазмоцитомы, низкий уровень парапротеина — IgG <50 г/л IgA <30 г/л BJ в моче <4 г/сут |
<0,6 (низкая) |
| II | Показатели средние между I и III стадиями | 0,6-1,2 (средняя) |
| III | Один или более из перечисленных: гемоглобин <85 г/л, уровень кальция сыворотки >3 ммоль/л, более 3 литических очагов в костях, высокий уровень парапротеина — IgG >70 г/л IgA >50 г/л BJ в моче >12 г/сут |
>1,2 (высокая) |
Дополнительным признаком, определяющим подстадию, является состояние функции почек:
А = относительно нормальная (креатинин сыворотки менее 170 мкмоль/л).
В = сниженная (креатинин сыворотки равен или более 170 мкмоль/л).
Прогноз и прогностические факторы
Множественная миелома — неизлечимое заболевание с медианой выживаемости 3 года. Только 10% больных может пережить 10-летний рубеж. Прогноз заболевания в значительной степени определяется опухолевой массой, о которой косвенно можно судить по стадии заболевания. При установлении I стадии медиана выживаемости превышает 60 мес, II — составляет 41 мес, III — достигает лишь 23 мес. Почечная недостаточность существенно ухудшает прогноз множественной миеломы, если не удается купировать ее симптомы в течение первых месяцев лечения.
К факторам неблагоприятного прогноза при диагностике множественной миеломы также относятся:
— высокий уровень бета2-микроглобулина;
— cнижение уровня альбумина;
— высокий пролиферативный индекс плазматических клеток (LI — labeling index);
— повышение ЛДГ;
— высокий уровень С-реактивного белка;
— цитогенетические аномалии:
— делеция или моносомия 13 хромосомы,
— делеция 17 хромосомы с мутацией или утратой гена-супрессора р53,
— множественные хромосомные аномалии,
— транслокации t(4;14)(p16,3;q32) и t(14;16)(q32;q23);
— возраст 69 лет и старше;
— общий статус по ECOG 3 и более.
Следует иметь в виду, что уровень бета2-микроглобулина является отражением массы опухоли, а ряд факторов свидетельствует о биологической активности опухоли: высокий пролиферативный индекс плазматических клеток, повышение ЛДГ, высокий уровень С-реактивного белка. По уровню С-реактивного белка можно судить об активности ИЛ–6 — основного фактора роста миеломы.
Лечение
После установления диагноза миеломы противоопухолевое лечение не во всех случаях необходимо начинать немедленно. При тлеющей и вялотекущей миеломе от химиотерапии следует воздержаться, поскольку срок от установления диагноза до появления первых клинических симптомов болезни может составить 5–8 лет. Больные с бессимптомной миеломой должны наблюдаться гематологом. Анализы крови и мочи, включая количественное определение уровня парапротеина, проводят каждые 3 мес.
Основанием для начала противоопухолевого лечения является появление признаков прогрессирования заболевания — повышение уровня сывороточного и/или мочевого парапротеина; появление болей в костях, связанных с костными поражениями; обнаружение мягкотканной плазмоцитомы, развитие анемии, гиперкальциемии или почечной недостаточности. Склонность к инфекциям при множественной миеломе также является показанием к началу цитостатического лечения.
Прежде чем говорить о современных возможностях лечения множественной миеломы, необходимо остановиться на оценке результатов терапии. Критерии оценки непосредственных результатов лечения при множественной миеломе представлены в таблице 3. Они основаны на представлениях о корреляции между уровнем парапротеина, массой опухоли и клиническими проявлениями.
Таблица 3
Критерии эффективности лечения множественной миеломы (SWOG, 1972; MTF, 1973)
| Степень ответа | Снижение уровня парапротеина |
| Полная ремиссия | На 75% и более в сыворотке крови и/или на 90% и более в моче |
| Истинная полная ремиссия* | На 100% в сыворотке крови и моче |
| Частичная ремиссия | На 50% и более, но менее 75% |
| Стабилизация | На 25% и более, но менее 50% |
| Отсутствие эффекта | Менее чем 25% |
Дополнительные клинические критерии ответа: гемоглобин 9 г/л; альбумин 30 г/л; нормальный уровень кальция, отсутствие появления новых литических очагов в костях.
*При истинной полной ремиссии число плазматических клеток в миелограмме не превышает 5%
Применение ВДХ для лечения множественной миеломы позволило реально повысить возможности достижения полных ремиссий. Это послужило основанием для внесения дополнений в общепринятые критерии оценки эффективности терапии. Так был введен новый критерий оценки — “истинная полная ремиссия”. Истинная полная ремиссия характеризуется полным исчезновением парапротеина из сыворотки крови и мочи, доказанным методом иммунофиксации, числом плазматических клеток в костном мозге менее 5% и отсутствием клинических проявлений заболевания. Любой эффект должен быть подтвержден дважды на протяжении ближайших 2 мес.
Оценка результатов лечения затруднена при низком уровне парапротеина и несекретирующей миеломе, а также в терминальной стадии болезни в связи с возможной утратой способности миеломных клеток к секреции парапротеина. В этих случаях необходимо ориентироваться на дополнительные клинические критерии ответа (табл. 3), выраженность болевого синдрома, общее состояние больных, динамику рентгенологических изменений в костях.
В последнее время при оценке эффективности лечения все шире используется понятие “фаза плато”, характеризующее стойкость ответа. Фаза плато устанавливается при стабилизации болезни, продолжающейся не менее 6 мес, с минимально выраженными клиническими проявлениями, с максимально низким уровнем парапротеина в сыворотке крови и/или моче, и отсутствием необходимости в гемотрансфузиях. Достижение фазы плато важно при решении вопроса о прекращении лечения.
Современное лечение множественной миеломы предполагает использование цитостатических препаратов (химиотерапия), лучевого и хирургического воздействий на опухоль. Кроме того, важное место в комплексе лечебных мероприятий занимают: глюкокортикоиды, бисфосфонаты, колониестимулирующие факторы (Г-КСФ, ГМ-КСФ), эритропоэтин (эпоэтин альфа, эпоэтин бета), интерферон альфа, а также методы экстракорпоральной детоксикации (плазмаферез, гемодиализ).
Поскольку множественная миелома представляет собой генерализованную плазмоклеточную опухоль, основным методом лечения первичных больных является химиотерапия. Наиболее широкое признание получили алкилирующие агенты (мелфалан, сарколизин, циклофосфамид, препараты нитрозомочевины) и их сочетания с преднизолоном. Стандартной терапией 1-й линии является сочетание мелфалана с преднизолоном (схема МР).
Схема МР:
I вариант: мелфалан — по 0,25 мг/кг (9мг/м2 ) в сутки с 1 по 4 день, внутрь, натощак; преднизолон — по 1–2 мг/кг/сут с 1 по 4 день, внутрь, после еды.
II вариант: мелфалан, в суммарной дозе 1 мг/кг за 5–7 дней, внутрь, натощак; преднизолон — по 1–2 мг/кг/сут в течение 5–7 дней, внутрь, после еды.
Курсы повторяются каждые 4–6 нед.
Поскольку биодоступность мелфалана при приеме внутрь составляет около 50%, ряд авторов рекомендует вводить его в/в из расчета 16 мг/м2 1 раз каждые 2 нед. Обычно удается провести 2–3 введения. Интервал между курсами составляет 4–6 нед. Во избежание выраженного угнетения нормального кроветворения при почечной недостаточности дозы мелфалана должны быть снижены на 30–50%. Общая эффективность программы МР составляет 50–60%, при медиане выживаемости 2–3 г.
Равноценное с мелфаланом действие оказывает циклофосфамид. Препарат менее гемодепрессивный, чем мелфалан и потому он чаще назначается больным с лейко— и/или тромбоцитопенией. Как правило, циклофосфамид используется в сочетании с преднизолоном (схема СР).
Схема СР:
циклофосфамид — по 400 мг через день до суммарной дозы 6–10 г, в/м;
преднизолон — 1мг/кг/сут с 1 по 7 день с постепенным снижением дозы, внутрь, после еды.
Курсы повторяются каждые 3–4 нед.
При назначении полихимиотерапии ремиссии достигаются чаще и быстрее, чем при использовании схемы МР. Однако на длительность ремиссии и продолжительность жизни это влияет мало.
Полихимиотерапия назначается при быстро прогрессирующей миеломе с исходными неблагоприятными прогностическими факторами или при развитии резистентности к терапии МР. Из схем полихимиотерапии (табл. 4) наиболее широкое распространение получили программы М2 и альтернирующий режим VMCP/VBAP.
Программа АВСМ предпочтительна при лечении пациентов с сопутствующим сахарным диабетом, язвенной болезнью желудка или 12ПК, а также винкристиновой нейропатией, поскольку схема не включает глюкокортикоидов и винкристина.
Схема VAD чаще используется при агрессивно протекающей миеломе, когда необходимо быстро получить ответ, а также при плазмоклеточном лейкозе. Эта программа наиболее показана при почечной недостаточности, когда трудно подобрать негемодепрессивную дозу мелфалана. Дозы вводимых препаратов редуцировать не следует. В последнее десятилетие программа VAD широко используется на индукционном этапе перед ВДХ.
Таблица 4
Основные схемы полихимиотерапии и гормонотерапии при множественной миеломе
| Схема | Лекарственное средство, доза и режим лечения |
| VMCP | Винкристин — по 1 мг/м2 в/в в 1 день, Мелфалан — по 6 мг/м2 внутрь с 1 по 4 день, Циклофосфамид — по 125 мг/м2 в/в с 1 по 4 день, Преднизолон — по 60 мг/м2 внутрь с 1 по 4 день. Курс повторяется каждый 21-28 день. |
| VBAP | Винкристин — по 1 мг/м2 в/в в 1 день, Кармустин* — по 30 мг/м2 в/в в 1 день, Доксорубицин — по 30 мг/м2 в/в в 1 день, Преднизолон — по 60 мг/м2 внутрь с 1 по 4 дни. Курс повторяется каждый 21-28 день. |
| VBMCP (M2) | Винкристин — по 1,2 мг/м2 в/в в 1 день, Кармустин* — по 20 мг/м2 в/в в 1 день, Мелфалан — по 8 мг/м2 внутрь с 1 по 4 день, Циклофосфамид — по 400 мг/м2 в/в в 1 день, Преднизолон — по 40 мг/м2внутрь с 1 по 7 день. Курс повторяется каждый 35-42 день. |
| ABCM | Доксорубицин — по 30 мг/м2 в/в в 1 день, Кармустин* — по 30 мг/м2 в/в в 1 день, Циклофосфамид — по 100 мг/м2 в/в с 22 по 25 день, Мелфалан — по 6 мг/м2 внутрь с 22 по 25 день. Курс повторяется каждый 46 день. |
| VAD | Винкристин — по 0,2 мг/м2 в режиме непрерывной в/в инфузии с 1 по 4 день, Доксорубицин — по 9 мг/м2 в режиме непрерывной в/в инфузии с 1 по 4 день, Дексаметазон — по 40 мг внутрь с 1 по 4 день, с 9 по 12 день, с 17 по 20 день. Курс повторяется каждый 28 день. |
| Дексаметазон | Дексаметазон — по 40 мг внутрь с 1 по 4 день, с 9 по 12 день, с 17 по 20 день. Курс повторяется каждый 28-35 день. |
| VID | Винкристин — по 1 мг/м2 в/в в 1 день, Идарубицин — по 10 мг/м2 внутрь с 1 по 4 день, Дексаметазон — по 40 мг внутрь с 1 по 4 день, с 9 по 12 день, с 17 по 20 день. Курс повторяется каждый 28 день. |
| NOP | Винкристин — по 2 мг в/в в 1 день, Митоксантрон — по 16 мг/м2 в/в в 1 день, Преднизолон — по 250 мг внутрь с 1 по 4 день, с 17 по 20 день. Курс повторяется каждый 28 день. |
| EDAP | Этопозид — по 100 мг/м2 в режиме непрерывной в/в инфузии с 1 по 4 день, Дексаметазон — по 40 мг внутрь с 1 по 5 день, Цитарабин — по 1 г/м2 в/в в 5 день, Цисплатин — по 25 мг/м2 в режиме непрерывной в/в инфузии с 1 по 4 день, ГМ-КСФ или Г-КСФ — по 250 мкг/м2 п/к c 6 дня. Курс повторяется после восстановления показателей крови. |
| DC-IE | Дексаметазон — по 40 мг внутрь с 1 по 4 день, Циклофосфамид — по 500 мг/м2 в/в в 5 день, Идарубицин — по 5 мг/м2 в/в с 8 по 10 день, Этопозид — по 100 мг/м2 в режиме непрерывной в/в инфузии с 8 по 10 день, ГМ-КСФ или Г-КСФ — по 250 мкг/м2 п/к с 12 по 20 день. Курс повторяется каждый 28 день или после восстановления показателей крови. |
*Кармустин может быть заменен ломустином в суточной дозе 80 мг внутрь в схемах VBAP и ABCM, VBMCP
Длительность химиотерапии зависит от времени достижения стойкой фазы плато. Как правило, фаза плато достигается после 6–12 курсов, после чего лечение продолжается еще 4–6 мес с последующей его отменой. Проведение поддерживающего лечения химиопрепаратами нецелесообразно из-за риска развития вторичного лейкоза и увеличения частоты инфекционных осложнений.
Обнадеживающие результаты по удлинению фазы плато получены при применении в этот период интерферона альфа в дозе 3 млн МЕ/м2 3 раза в неделю. Такое лечение проводится не менее 1 г. Поддерживающая терапия препаратами интерферона альфа–2б и интерферона альфа–2а увеличивает выживаемость, свободную от прогрессирования, на 4–6 мес, а общую — на 4–7 мес.
Препятствием для широкого использования интерферонов альфа являются их высокая стоимость, а также возможные осложнения (цитопении, гриппоподобный синдром, потеря веса, депрессия и др.). Вместе с тем во время лечения интерфероном альфа уменьшается частота инфекций. Наиболее целесообразно назначать интерферон альфа молодым больным множественной миеломой в фазе плато с малой опухолевой массой.
Рецидивы при множественной миеломе развиваются неизбежно у большинства больных. Если рецидив развивается в течение 6 мес после установления фазы плато (ранний рецидив), то возобновление первичной терапии вызывает повторный эффект только у половины больных. Эффективность возобновления первичной химиотерапии выше у пациентов с длительностью фазы плато более 1 года.
В случае развития ранних рецидивов целесообразна смена программы лечения. Одной из наиболее эффективных в таких случаях является схема VAD. Частота достижения объективных ответов составляет 40–65%. Кроме того, для лечения рецидивов используются программы, аналогичные VAD: VID, NOP. В схеме NOP замена доксорубицина на митоксантрон уменьшает кардиотоксичность. В схеме VID идарубицин с удлиненным периодом полувыведения (T1/2), для приема внутрь, используется как альтернатива в/в инфузии доксорубицина. Возможно также применение высоких доз кортикостероидов в монорежиме, особенно у больных с миелосупрессией. Резистентность к программе VAD и ее аналогам является признаком полирезистентности опухоли к химиопрепаратам.
Для преодоления лекарственной резистентности используются программы EDAP, DC-IE, а также циклофосфамид в высокой дозе 3,6 г/м2. Введение высоких доз циклофосфамида обязательно сопровождается введением месны.
В последние несколько лет для преодоления лекарственной резистентности при миеломе используется Талидомид, препарат широко применявшийся в 50-е годы прошлого столетия как седативное и снотворное средство, а затем запрещенный в 1961 г. из-за тератогенного эффекта. Препарат в России не зарегистрирован. Талидомид оказывает антиангиогенное действие, индуцирует апоптоз миеломных клеток, ингибирует экспрессию цитокинов, участвующих в регуляции пролиферации опухолевых клеток.
Талидомид назначается по 200 мг/сут внутрь, при хорошей переносимости препарата через 2 нед доза увеличивается до 400 мг/сут. В настоящее время имеются убедительные данные, свидетельствующие о высокой эффективности сочетания низких доз Талидомида с дексаметазоном. При сочетании 2 препаратов Талидомид назначается по 100 мг/сут длительно, а дексаметазон — по 20 мг/м2 внутрь в течение первых 4 дней каждого месяца.
Из побочных эффектов Талидомида следует обратить внимание на сонливость, нейротоксичность, отеки, запоры, слабость, головокружения, тромбозы глубоких вен.
Неудовлетворенность результатами стандартной химиотерапии и отсутствие принципиально новых противоопухолевых средств явилось стимулом к разработке программ ВДХ с последующей трансплантацией гемопоэтических стволовых клеток. Исследования, проведенные в течение последних 2 десятилетий, свидетельствуют о высокой эффективности и безопасности использования высоких доз мелфалана (140 мг/м2 или 200 мг/м2) с последующей трансплантацией аутологичных периферических стволовых клеток крови или костного мозга. ВДХ мелфаланом (140 мг/м2) позволяет получить полные ремиссии у 30–50% первичных больных множественной миеломой, смертность из-за токсических осложнений при этом не превышает 5%.
Французской группой по изучению множественной миеломы (IFM) в крупном рандомизированном исследовании (IFM90) было показано преимущество ВДХ мелфаланом перед стандартной полихимиотерапией (VMCP/VBAP). При ВДХ частота достижения полных и истинных полных ремиссий была значительно выше (38%), чем при использовании стандартной полихимиотерапии (14%). Длительное наблюдение за больными показало, что высокодозное лечение улучшает и отдаленные результаты. Так, 7-летняя общая выживаемость оказалась выше (43 против 25%) при высокодозной терапии, как и медиана выживаемости (57 против 44 мес). Высокодозное лечение улучшило и бессобытийную выживаемость. 7-летняя бессобытийная выживаемость составила 16% после высокодозного лечения против 8% в контрольной группе, а медиана бессобытийной выживаемости — 28 по сравнению с 18 мес.
В трех крупных исследованиях, где проводилось сравнение результатов высокодозной и стандартной химиотерапии (исторический контроль), было подтверждено, что высокодозное лечение с аутотрансплантацией гемопоэтических стволовых клеток (ГСК) улучшает как непосредственные, так и отдаленные результаты. Высокодозное лечение увеличивает медиану общей выживаемости на 14 мес и более.
C целью улучшения результатов высокодозного лечения проводятся исследования по оценке различных режимов кондиционирования. Повышение дозы мелфалана со 140 до 200 мг/м2 позволило увеличить частоту полных ремиссий до 70%, что, как правило, улучшает и показатели выживаемости. Этот режим кондиционирования оказался менее токсичным, чем сочетание мелфалана в дозе 140 мг/м2 с облучением всего тела. Поэтому при проведении ВДХ с последующей аутологичной трансплантацией ГСК более обосновано кондиционирование мелфаланом в дозе 200 мг/м2.
Другой подход, позволивший увеличить частоту полных ремиссий при множественной миеломе, заключается в проведении повторных курсов ВДХ с трансплантацией ГСК. Исследовательской группой из Арканзаса было показано, что повторная высокодозная терапия с тандемной трансплантацией ГСК увеличивает частоту полных ремиссий с 24 до 43%.
В рандомизированном исследовании (IFM94), проведенном французской группой по изучению множественной миеломы, было установлено, что 2-кратная ВДХ с аутотрансплантацией ГСК улучшает отдаленные результаты лечения. Общая 7-летняя выживаемость составила после первой ВДХ 21 и 42% после 2 курсов с тандемной трансплантацией. 7-летняя бессобытийная выживаемость также была значительно выше после повторной ВДХ в сравнении с однократной (20 против 10%).
Исследования по сравнению эффективности 2-кратной высокодозной терапии с аутотрансплантацией ГСК и стандартной химиотерапии (исторический контроль) проведены исследовательской группой из Арканзаса. Результаты этого исследования показали, что независимо от прогностических факторов общая выживаемость больных множественной миеломой после 2-кратной ВДХ с тандемной трансплантацией ГСК значительно лучше, чем при использовании химиотерапии в стандартных дозах.
При 2-кратной высокодозной терапии с аутотрансплантацией ГСК медиана общей выживаемости не достигнута после 9 лет наблюдения в группе больных с благоприятным прогнозом и составила 4,8 года в группе больных с неблагоприятными прогностическими признаками. При использовании стандартной химиотерапии медиана общей выживаемости во всей группе больных составила 3,6 года.
Опыт использования высокодозной терапии с аутотрансплантацией ГСК при множественной миеломе позволил выделить несколько прогностических факторов, влияющих на результаты лечения. К основным факторам благоприятного прогноза относятся: отсутствие моносомии или делеции 13 хромосомы, низкий уровень С-реактивного белка (< 4 мг/л) и бета2-микроглобулина (< или = 4 мг/л), отсутствие предшествующего лечения или использование стандартной химиотерапии менее 1 года. Результаты высокодозного лечения хуже при А-миеломе.
Современные подходы к лечению множественной миеломы предполагают раннее использование 2-кратной ВДХ с аутотрансплантацией периферических ГСК. ВДХ обычно рекомендуется для лечения первичных больных моложе 60 лет.
Режим 2-кратной ВДХ с аутотрансплантацией ГСК включает:
1. Индукционная терапия — 3–4 курса полихимиотерапии по схеме VAD.
2. Мобилизация периферических ГСК: циклофосфамид — по 4–6 г/м2, в/в, с последующим назначением препаратов колониестимулирующих факторов (Г-КСФ или ГМ-КСФ) из расчета 250 мкг/м2, п/к, в течение 6–14 дней в зависимости от сроков восстановления гемопоза*.
3. Сбор периферических ГСК (CD34+) не менее 4·106/кг методом повторных цитаферезов.
4. ВДХ — мелфалан 200 мг/м2, в/в (кондиционирование)*.
5. В/в введение периферических ГСК (CD34+) не менее 2·106/кг.
6. Повторная ВДХ мелфаланом (через 3–6 мес) с последующим в/в введением периферических ГСК (CD34+) 2·106/кг и более.
Аутотрансплантация ГСК осуществляется после каждого введения мелфалана, потому она и называется тандемной.
* Для мобилизации периферических ГСК и кондиционирования используются и другие режимы.
Серьезную проблему представляет лечение рецидивов после ВДХ. В таких случаях чаще используется программа полихимиотерапии по схеме DCEP.
DCEP:
Дексаметазон — по 40 мг внутрь с 1 по 4 день, цисплатин — по 15 мг/м2/сут, постоянное в/в введение с помощью инфузомата с 1 по 4 день, циклофосфамид — по 300 мг/м2/сут, постоянное в/в введение с помощью инфузомата с 1 по 4 день, этопозид — 30 мг/м2/сут, постоянное в/в введение с помощью инфузомата с 1 по 4 день, Г-КСФ — по 300 мкг/сут п/к до восстановления числа гранулоцитов.
Совершенно очевидно, что высокодозное лечение при множественной миеломе не предотвращает развития рецидивов. С целью контроля минимальной остаточной болезни и для поддержания ремиссии после высокодозной терапии назначается интерферон альфа. Результаты рандомизированного исследования по оценке эффективности поддерживающей терапии интерфероном альфа после высокодозного лечения показали, что его назначение в дозе 3 млн МЕ/м2 3 раза в неделю удлиняет медиану выживаемости, свободной от прогрессирования, до 42 мес по сравнению с 27 мес в группе без использования интерферона альфа.
Необходимо отметить, что назначение интерферона альфа после высокодозной терапии увеличивает общую выживаемость почти на 2,5 года.
Последние годы разрабатываются принципиально новые подходы контроля резидуальной опухоли. Определенные надежды возлагаются на использование моноклональных антител к ИЛ–6. Имеются данные о том, что нейтрализация продукции этого цитокина после ВДХ с помощью моноклональных антител В-Е8 препятствует развитию рецидива. Проводятся исследования по оценке эффективности применения специальных вакцин (идиотипических или ДНК-вакцин) или дендритических клеток, представляющих идиотип определенного миеломного белка, усиливающих иммунный противоопухолевый ответ собственных Т-лимфоцитов.
С 1983 г. для лечения множественной миеломы применяется ВДХ с аллоТКМ. Преимущество этого метода перед аутологичной трансплантацией ГСК заключается в отсутствии риска контаминации трансплантата миеломными клетками. Кроме того, доказано противоопухолевое действие самой аллогенной трансплантации, благодаря эффекту “трансплантат против миеломы”.
Двадцатилетний мировой опыт применения ВДХ с аллогенной трансплантацией ГСК свидетельствует о высокой эффективности метода, позволяющего получить полные ремиссии у 30–60% больных множественной миеломой.
Следует обратить внимание на то, что у половины больных с полной ремиссией она доказана с помощью молекулярно-биологических методов исследования (молекулярная ремиссия). При ВДХ с аутологичной трансплантацией костного мозга молекулярные ремиссии являются большой редкостью.
Аллогенная трансплантация ГСК выполняется только пациентам моложе 50 лет, имеющим HLA идентичного сиблинга. Реально ее удается произвести лишь 5–10% больных миеломой. Смертность остается очень высокой, главным образом из-за инфекционных осложнений и развития болезни “трансплантат против хозяина”. Необходимо отметить, что по данным Европейской группы по трансплантации ГСК крови и костного мозга (EBMT), в последнее десятилетие отмечается уменьшение смертности с 38 до 21% в течение 6 мес после трансплантации. Это связано с внедрением новых эффективных антибактериальных, противогрибковых и противовирусных средств. Медиана общей выживаемости также увеличилась с 10 до 50 мес.
Исследования Европейской группы EBTM показали отсутствие преимущества аллогенной трансплантации ГСК перед аутологичной. Рецидивы после достижения полной гематологической ремиссии возникают реже после аллогенной трансплантации ГСК, чем после аутологичной. Однако общая выживаемость выше после аутологичной трансплантации за счет низкой летальности, связанной с трансплантацией.
Интересные данные были получены при выполнении ВДХ с аллогенной трансплантацией ГСК в начальной стадии множественной миеломы. У одной трети пациентов, достигших полной ремиссии после АллоТКМ, отсутствуют признаки прогрессирования болезни после 6 лет наблюдения. Не исключено, что единственным радикальным методом лечения множественной миеломы может стать ВДХ с аллогенной ГСК за счет противоопухолевого эффекта “трансплантат против миеломы”.
Аллогенная трансплантация редко используется для лечения первичных больных множественной миеломой из-за высокой частоты летальных осложнений этого метода. Согласно рекомендациям IX Международного совещания по множественной миеломе 2003 г., ВДХ с аллогенной трансплантацией ГСК может быть предложена молодым больным, имеющим HLA-cовместимого сиблинга того же пола, при отсутствии инфицированности цитомегаловирусом.
Определенный интерес представляет разрабатываемый в последнее время метод аллогенной трансплантации ГСК крови или костного мозга после немиелоаблативных режимов кондиционирования, так называемая мини-аллогенная трансплантация. Цель этого метода заключается в достижении эффекта “трансплантат против миеломы” в условиях меньшей токсичности, благодаря использованию немиелоаблативных режимов химиотерапии.
Наиболее интересной представляется идея использования мини-аллогенной трансплантации после ВДХ с последующей аутологичной трансплантацией (тандемная ауто/аллогенная трансплантация) для более полного контроля остаточной опухоли.
Как плазмоцитома, так и множественная миелома относятся к высокорадиочувствительным опухолям. Тем не менее лучевая терапия как самостоятельный метод используется крайне редко — только при плазмоцитомах с полным отсутствием М-компонента. Для лечения костных и экстрамедулярных мягкотканных плазмоцитом суммарная доза лучевой терапии составляет 40–50 Гр. Эффективность лечения выше у больных с поражением мягких тканей в сравнении с костными плазмоцитомами. При мягкотканных плазмоцитомах рецидивы развиваются приблизительно у 20–25% больных, генерализация с развитием множественной миеломы наблюдается редко. При костных плазмоцитомах в течение 10 лет в 55% случаев развивается типичная миелома.
При множественной миеломе локальная лучевая терапия проводится в суммарной дозе 30–40 Гр (РОД — 2 Гр) при угрозе патологических переломов в опорных частях скелета на крупные очаги поражения (позвоночник, кости таза, бедренные, мало-и большеберцовые, плечевые кости). Такие же дозы лучевой терапии используются при неврологической симптоматике, связанной со сдавлением опухолью спинного мозга или его корешков.
Лучевая терапия может быть назначена в качестве паллиативной меры при сильных локальных болях в костях, особенно при развитии резистентности к химиотерапии в терминальной стадии болезни. Обычно в этой ситуации суммарная доза облучения составляет 30 Гр (РОД — 3 Гр).
Успех лечения множественной миеломы зависит не только от выбора оптимального метода противоопухолевой терапии, но и от эффективности борьбы с многочисленными осложнениями.
Лечение инфекционных осложнений проводится в соответствии с общими принципами лечения больных с иммунодефицитом. В последние годы в связи с интенсификацией программ химиотерапии глубокая нейтропения у больных множественной миеломой развивается довольно часто. В период нейтропении инфекции могут протекать атипично. Лихорадка может быть единственным симптомом инфекции, в связи с чем необходимо назначение антибиотиков широкого спектра действия. При выборе антибактериального препарата следует избегать назначения нефротоксичных аминогликозидов: гентамицина, канамицина, стрептомицина и др. Одновременно с антибактериальной назначается противогрибковая терапия (флуконазол, кетоконазол и др.).
С целью профилактики инфекционных осложнений используются препараты иммуноглобулина для в/в введения в дозе 0,4 мг/кг 1 раз в месяц. На фоне химиотерапии, особенно во время первых курсов лечения, с профилактической целью можно использовать ко-тримоксазол, ципрофлоксацин.
Грибковые инфекции часто развиваются при лечении дексаметазоном в высоких дозах или другими глюкокортикоидами. Риск развития грибковых инфекций также увеличивается в период нейтропении после высокодозного лечения. С профилактической целью обычно используются флуконазол, кетоконазол. Основным препаратом для лечения грибковых инфекций является амфотерицин В, но при его назначении необходим тщательный контроль функции почек и соответствующая коррекция дозы препарата при появлении признаков нефротоксичности.
Профилактика и лечение почечной недостаточности при множественной миеломе занимают особое место. С целью своевременной профилактики развития острой почечной недостаточности необходимо снижение гиперкальциемии, назначение аллопуринола во время первых курсов химиотерапии, особенно при большой массе опухоли. Особое внимание необходимо уделять своевременной диагностике, а также лечению мочевой инфекции. Кроме того, перед началом химиотерапии, даже при минимальных признаках почечной недостаточности, необходима предварительная гидратация. Гидратация также необходима во время инфекции в связи с опасностью развития острой почечной недостаточности.
Основными методами лечения почечной недостаточности, обусловленной миеломой, являются адекватная химиотерапия и гидратация. Следует учитывать тот факт, что мелфалан не является нефротоксичным препаратом. Тем не менее больным множественной миеломой, страдающим почечной недостаточностью, его необходимо назначать в сниженных дозах из-за повышения риска развития миелосупрессии, обусловленной нарушением выведения препарата почками. При почечной недостаточности следует отдавать предпочтение программе VAD. Схема VAD отличается малой гематологической токсичностью и быстрым противоопухолевым действием. Почечная недостаточность может быть обратимой при адекватной гидратации и химиотерапии. При тяжелой уремии показан гемодиализ или использование других методов экстракорпоральной детоксикации. Ряд авторов предлагают с целью лечения почечной недостаточности больным множественной миеломой проводить плазмаферез.
Плазмаферез также используется при синдроме гипервязкости с кровоточивостью, когда уровень белка сыворотки крови превышает 130–140 г/л. Абсолютным показанием к проведению плазмафереза является парапротеинемическая кома.
Основными методами лечения гиперкальциемии является химиотерапия и глюкокортикостероиды. Дополнительную роль играет гидратация больных. Выбор лечебной тактики зависит от своевременности диагностики и определения степени тяжести гиперкальциемии. При уровне кальция от 2,6 до 3,5 ммоль/л диагностируется легкая степень гиперкальциемии. Токсическая форма характеризуется содержанием кальция в сыворотке крови выше 3,5 ммоль/л. В этой ситуации необходимо немедленное начало лечения. Поскольку гиперкальциемия может сопровождаться развитием гипокалиемии, необходим контроль уровня калия и соответствующая его коррекция.
При гиперкальциемии легкой степени больной должен выпивать в сутки до 3 л минеральной воды. При токсической гиперкальциемии рекомендуется вводить в/в от 2–3 до 5 л изотонического раствора натрия хлорида и форсировать диурез в/в введениями фуросемида по 20–40 мг/сут.
Современная стандартная терапия гиперкальциемии включает в/в введение бисфосфонатов — препаратов, ингибирующих резорбцию кости остеокластами. Памидроновая кислота вводится в/в капельно в течение 2–4 ч в дозе от 30 до 90 мг в зависимости от уровня кальция сыворотки крови.
Золедроновая кислота назначается в дозе 4 мг и вводится в/в в течение 15 мин. Доза ибандроновой кислоты составляет 4–8 мг, препарат вводится в/в медленно.
При множественной миеломе бисфосфонаты также используются для профилактики гиперкальциемии и развития других костных осложнений. Лечение бисфосфонатами уменьшает интенсивность болей в костях, сдерживает распространение остеодеструкций и снижает частоту переломов костей. Бисфосфонаты следует назначать всем больным множественной миеломой, имеющим поражение костей.
Для профилактики развития костных осложнений при множественной миеломе назначается памидроновая кислота по 90 мг, а золедроновая кислота по 4 мг каждый месяц.
При патологических переломах длинных трубчатых костей необходима хорошая репозиция и фиксация отломков. Если переломы возникают в местах крупных опухолевых поражений с большим диастазом отломков, осуществляется остеосинтез. При компрессионных переломах позвонков, сопровождающихся корешковым болевым синдромом, применяются химиотерапия и аналгетики. Ношение корсета показано в редких случаях.
При компрессии спинного мозга и подтверждении ее опухолевой природы одним из основных методов лечения является локальная лучевая терапия. Дополнительно назначается дексаметазон в высоких дозах — 16 мг/сут. Другим вариантом лечения является интенсивная системная химиотерапия. Обычно используется схема VAD.
Анемия развивается у большинства больных множественной миеломой. Эффективная химиотерапия обычно сопровождается повышением уровня гемоглобина. При глубокой анемии необходимы трансфузии эритроцитной массы. Поддержание уровня гемоглобина выше 10 г/дл особенно важно у пожилых больных и пациентов с сопутствующими сердечно-сосудистыми заболеваниями. В последние годы для лечения анемии используется эритропоэтин. Рекомбинантный эритропоэтин эффективен у 60–65% больных множественной миеломой. Он назначается в дозе 150–200 МЕ/кг/сут 3 раза в неделю. Использование эритропоэтина избавляет от необходимости гемотрансфузий и позволяет избежать осложнений трансфузионной терапии.
Литература
Андреева Н.Е. и др. ...Руководство по гематологии/Под ред. А.И. Воробьева.— М.: Ньюдиамед, 2003.— Т. 2.— С. 151–172.
Вотякова О.М. и др. ...Клиническая онкогематология/Под ред. М.А. Волковой.— М.: Медицина, 2001.— C. 423–449.
Cancer: principles and practice of oncology/Ed. V.T. DeVita et al.— 6th ed.— Philadelphia — Baltimor — NY — London — Buenos Aires — Hong Kong — Sydney — Tokyo: Lippincott Williams and Wikins, 2001.— 3235 p.
Durie B.G.M. et al. Initial recomendations international myeloma working group. A publcation of International myeloma foundation.— 2003.— 30 p.
Harris N. et al. ...Blood.— 1994.— V. 84.— P. 1361–1392.
Multiple myeloma/ Ed. G. Garton et al. London — Sydney — Auckland: Arnold, 1996.— 219 p.
Myeloma: Biology and management/Ed. J.S. Malpas et al.— 2nd ed.— Oxford, NY, Tokyo: Oxford University Press, 1998.— 673 p.
Myeloma/ Ed. J. Mehta et al.— Martin Dunitz LTD, 2002.— 539 p.
World health organization classification of tumors. Pathology and genetics of tumors of haematopoietic and lymphoid tissues/Ed. E. Jaffe et al.— Lyon: IARC Press, 2001.— 351 p.
Описание проверено экспертом

Оцените статью:
- Ретикулоплазмоцитоз
- Рустицкого болезнь
- Рустицкого-Калера болезнь
- Миеломатоз
- Плазмоклеточная миелома
Полужирным шрифтом выделены лекарства, входящие в справочники текущего года. Рядом с названием препарата может быть указан ежегодный уровень индекса информационного спроса (показатель, который отражает степень интереса потребителей к информации о лекарстве).
События
Реклама
